
Immune Monitoring during Therapy Reveals Activitory and Regulatory Immune Responses in High-Risk Neuroblastoma

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
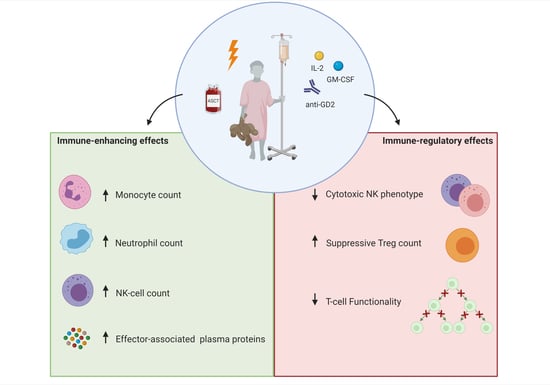
1. Introduction
2. Materials and Methods
2.1. Patients and Treatment
2.2. Sample Collection
2.3. Treg and NK-Cell Phenotyping
2.4. Proliferation Assay
2.5. Suppression Assay
2.6. Protein Profiling
2.7. Statistics
3. Results
3.1. Patient Characteristics
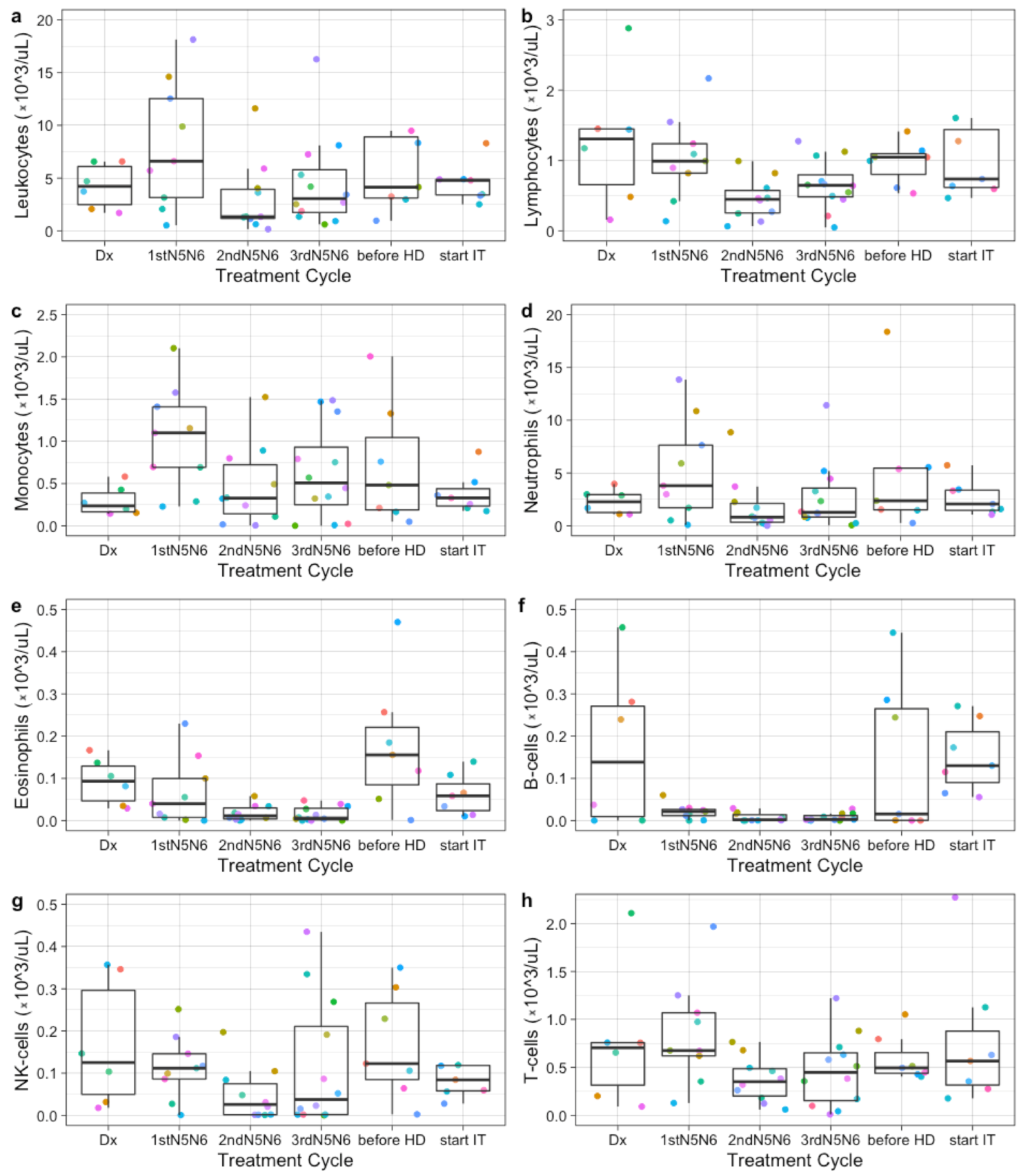
3.2. Immune Profiles at Diagnosis, during Induction Chemotherapy, and before High-Dose Chemotherapy Show Broad Variation between Patients
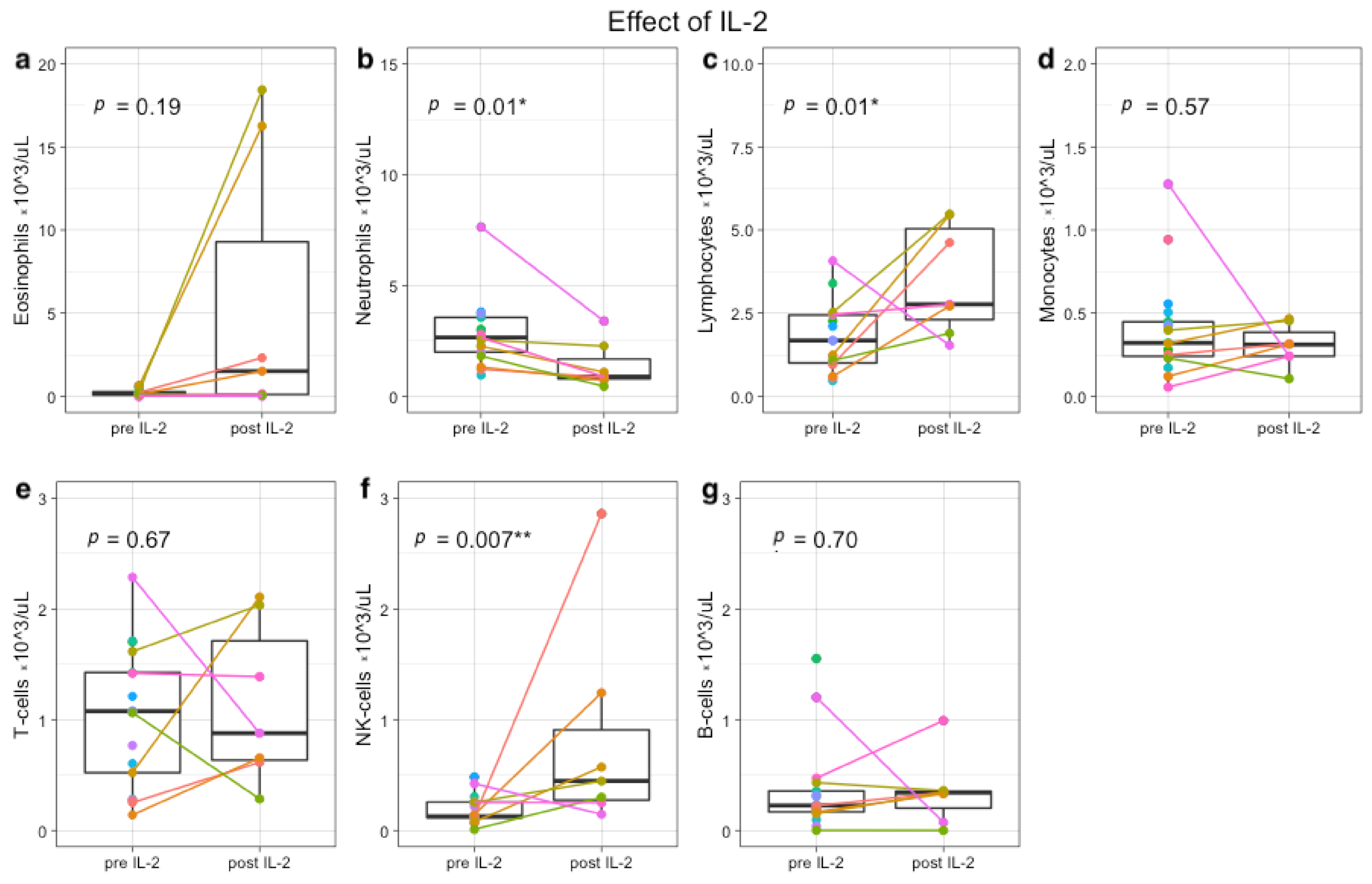
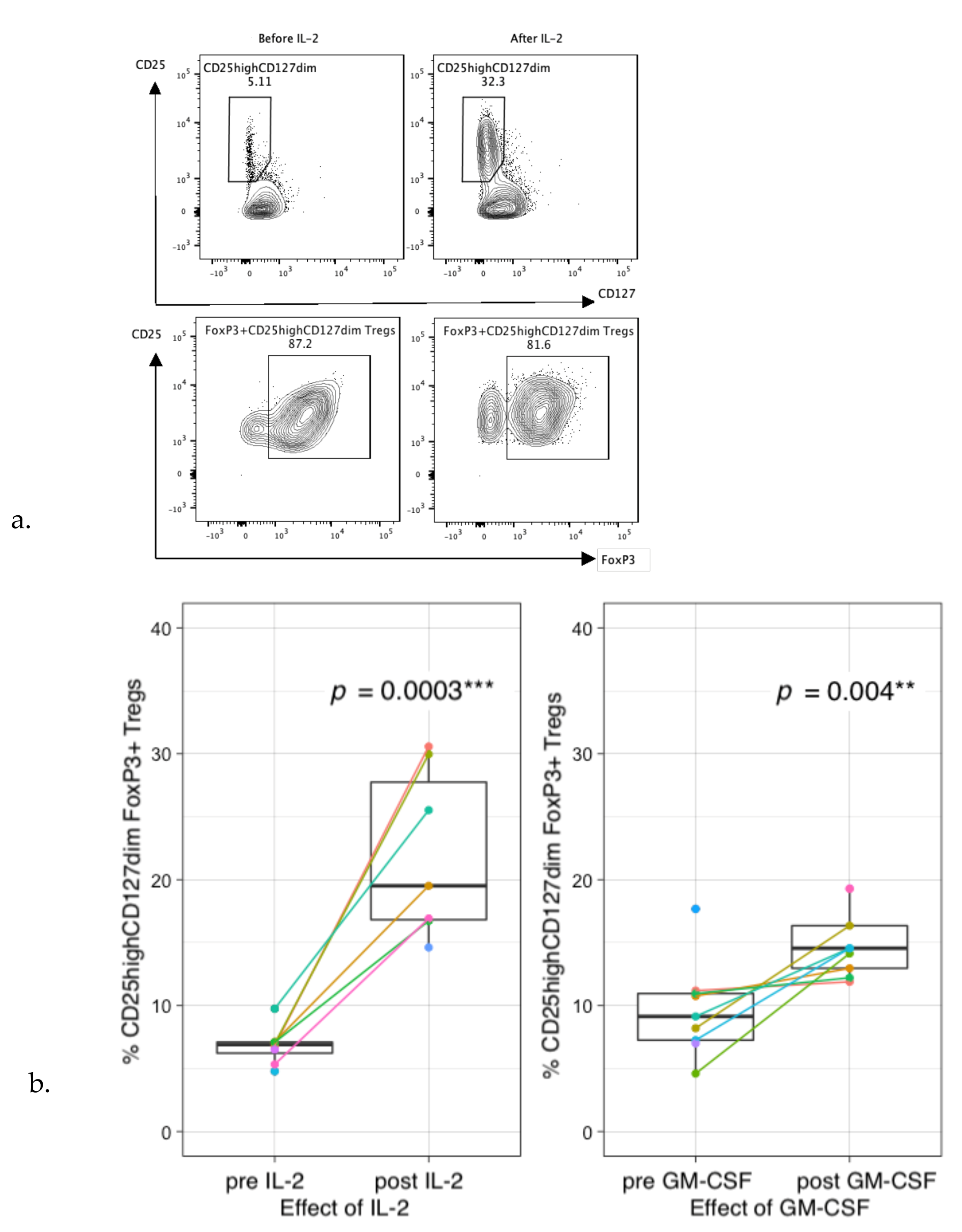
3.3. Immune Profiles during Immunotherapy Show Effect of IL-2 and GM-CSF on Leukocyte and Lymphocyte Subsets
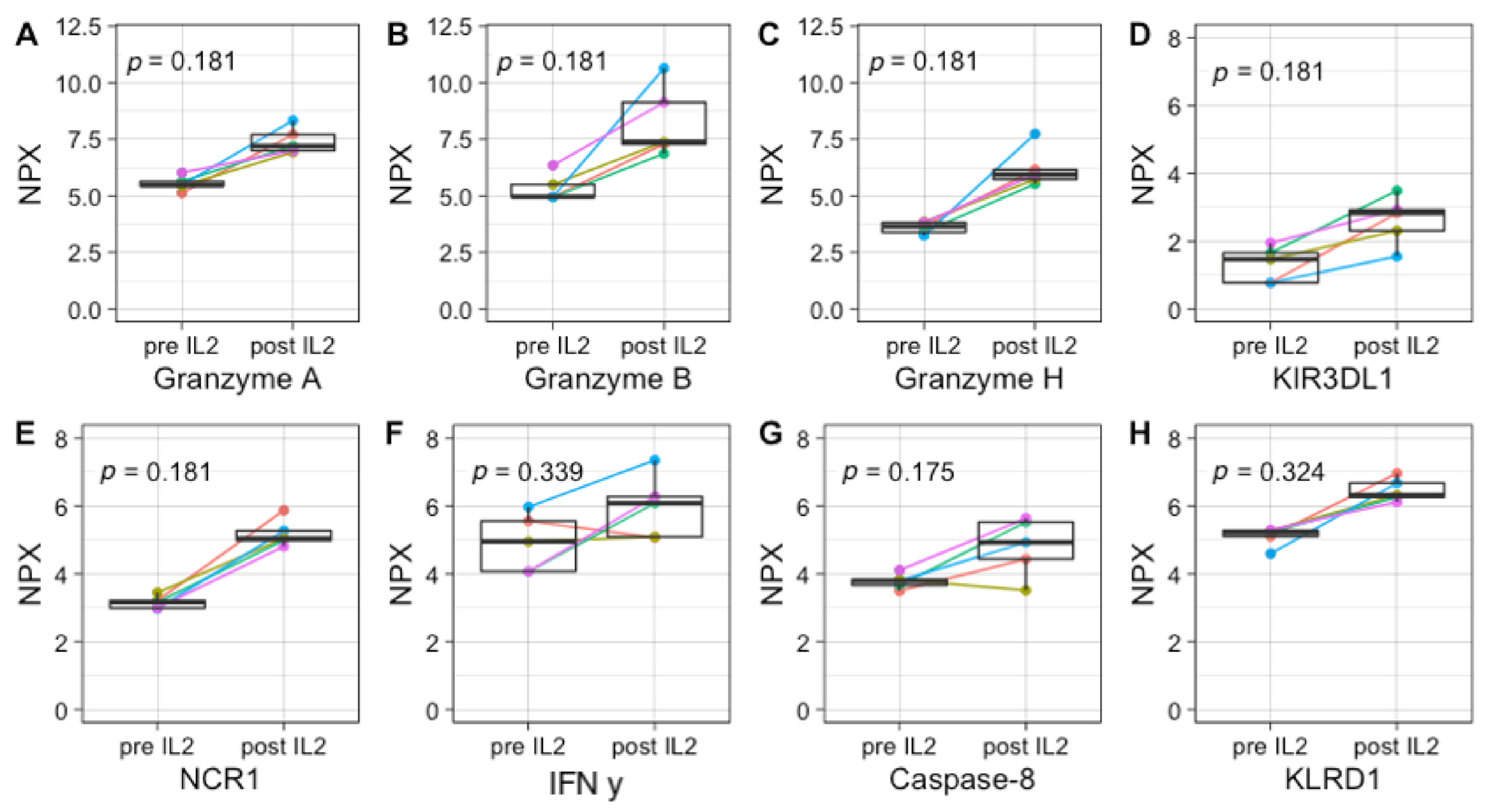
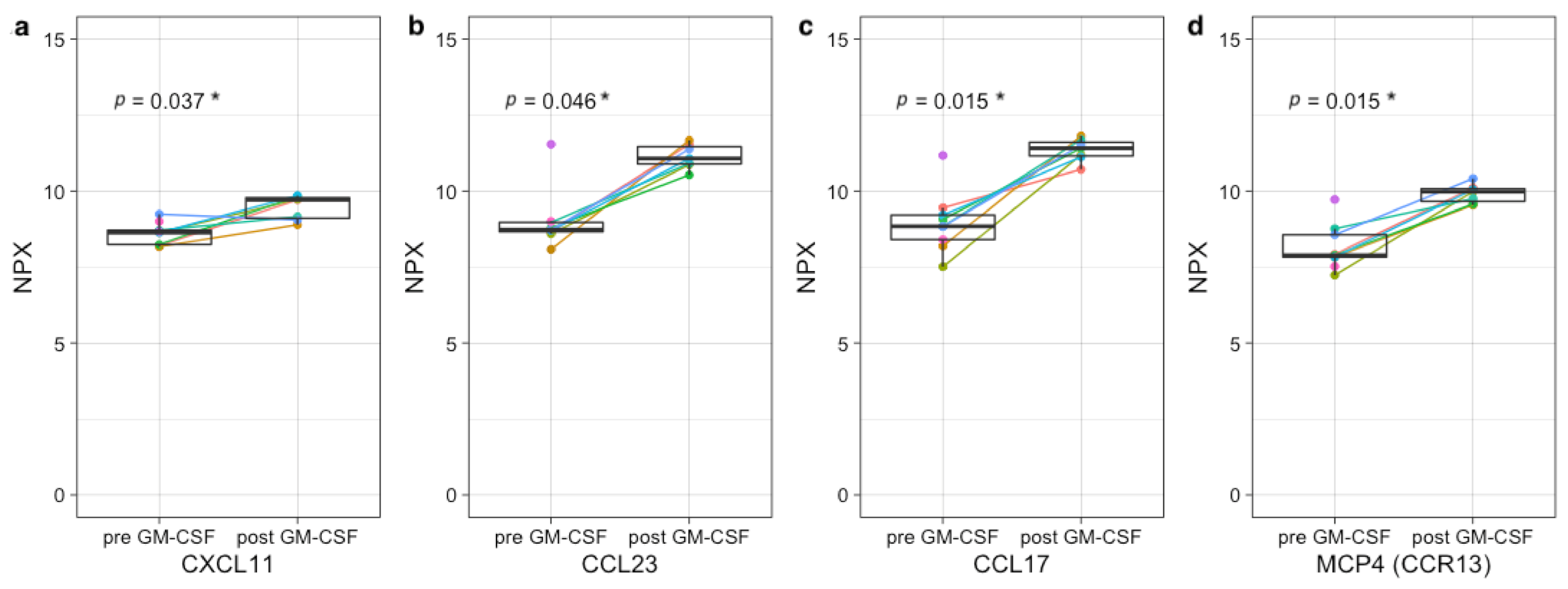
3.4. Plasma Protein Profiling Further Supports IL-2 and GM-CSF Mediated Immune Engagement during Immunotherapy
3.5. NK-Cell Phenotype Varies Widely between Patients and Is Suboptimal for Efficient Dinutuximab-Mediated Cytotoxicity
3.6. Preferential Treg Expansion and Impaired T-Cell Proliferation during Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2016. [Google Scholar]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladenstein, R.; Pötschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Yaniv, I.; Laureys, G.; Brock, P.; Michon, J.M.; Owens, C.; et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): A multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1617–1629. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; Naranjo, A.; Diccianni, M.B.; Gan, J.; Hank, J.A.; Batova, A.; London, W.B.; Tenney, S.C.; et al. Long-term follow-up of a Phase III Study of ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-risk Neuroblastoma: COG Study ANBL0032. Clin. Cancer Res. 2021, 18. [Google Scholar] [CrossRef]
- Park, J.R.; Bagatell, R.; London, W.B.; Maris, J.M.; Cohn, S.L.; Mattay, K.M.; Hogarty, M. Children’s Oncology Group’s 2013 blueprint for research: Neuroblastoma. Pediatr. Blood Cancer 2013, 60, 985–993. [Google Scholar] [CrossRef]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in risk classification and treatment strategies for neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef]
- Masucci, G.; Ragnhammar, P.; Wersäll, P.; Mellstedt, H. Granulocyte-monocyte colony-stimulating-factor augments the interleukin-2-induced cytotoxic activity of human lymphocytes in the absence and presence of mouse or chimeric monoclonal antibodies (mAb 17-1A). Cancer Immunol. Immunother. 1990, 31, 231–235. [Google Scholar] [CrossRef]
- Hank, J.A.; Surfus, J.; Sondel, P.M.; Robinson, R.R.; Mueller, B.M.; Reisfeld, R.A.; Cheung, N.K. Augmentation of Antibody Dependent Cell Mediated Cytotoxicity following in Vivo Therapy with Recombinant Interleukin 2. Cancer Res. 1990, 50, 5234–5239. [Google Scholar]
- Thoma, M.D.; Huneke, T.J.; DeCook, L.J.; Johnson, N.D.; Wiegand, R.A.; Litzow, M.R.; Hogan, W.J.; Porrata, L.F.; Holtan, S.G. Peripheral blood lymphocyte and monocyte recovery and survival in acute leukemia postmyeloablative allogeneic hematopoietic stem cell transplant. Biol. Blood Marrow Transplant. 2012, 18, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Galvez-Silva, J.; Maher, O.M.; Park, M.; Liu, D.; Hernandez, F.; Tewari, P.; Nieto, Y. Prognostic Analysis of Absolute Lymphocyte and Monocyte Counts after Autologous Stem Cell Transplantation in Children, Adolescents, and Young Adults with Refractory or Relapsed Hodgkin Lymphoma. Biol. Blood Marrow Transplant. 2017, 23, 1276–1281. [Google Scholar] [CrossRef]
- Kim, H.T.; Armand, P.; Frederick, D.; Andler, E.; Cutler, C.; Koreth, J.; Alyea, E.P.; Antin, J.H.; Soiffer, R.J.; Ritz, J.; et al. Absolute lymphocyte count recovery after allogeneic hematopoietic stem cell transplantation predicts clinical outcome. Biol. Blood Marrow Transplant. 2015, 21, 873–880. [Google Scholar] [CrossRef] [Green Version]
- Nassin, M.L.; Nicolaou, E.; Gurbuxani, S.; Cohn, S.L.; Cunningham, J.M.; LaBelle, J.L. Immune Reconstitution Following Autologous Stem Cell Transplantation in Patients with High-Risk Neuroblastoma at the Time of Immunotherapy. Biol. Blood Marrow Transplant. 2018, 24, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Scheid, C.; Pettengell, R.; Ghielmini, M.; Radford, J.A.; Morgenstern, G.R.; Stern, P.L.; Crowther, D. Time-course of the recovery of cellular immune function after high-dose chemotherapy and peripheral blood progenitor cell transplantation for high-grade non-Hodgkin’s lymphoma. Bone Marrow Transplant. 1995, 15, 901–906. [Google Scholar]
- Ye, C.; Brand, D.; Zheng, S.G. Targeting IL-2: An unexpected effect in treating immunological diseases. Signal Transduct. Target. Ther. 2018, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Dutch Childhood Oncology Group (DCOG). DCOG NBL 2009 Treatment Protocol for Risk Adapted Treatment of Children with Neuroblastoma Admendment 1; 2012. [Google Scholar]
- Brodeur, G.M.; Pritchard, J.; Berthold, F.; Carlsen, N.L.; Castel, V.; Castelberry, R.P.; De Bernardi, B.; Evans, A.E.; Favrot, M.; Hedborg, F. Revisions of the international criteria for neuroblastoma diagnosis, staging and response to treatment. Prog. Clin. Biol. Res. 1994, 385, 363–369. [Google Scholar] [CrossRef]
- De Jager, W.; Prakken, B.J.; Bijlsma, J.W.J.; Kuis, W.; Rijkers, G.T. Improved multiplex immunoassay performance in human plasma and synovial fluid following removal of interfering heterophilic antibodies. J. Immunol. Methods 2005, 300, 124–135. [Google Scholar] [CrossRef]
- Warnes, G.J.; Bolker, B.; Bonebakker, L.; Gentleman, G.; Liaw, W.H.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M.; et al. gplots: Various R Programming Tools for Plotting Data. R Package Version 2019, 2, 1. [Google Scholar]
- Url, S.; Society, R.S.; Society, R.S. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Angelo, L.S.; Banerjee, P.P.; Monaco-Shawver, L.; Rosen, J.B.; Makedonas, G.; Forbes, L.R.; Mace, E.M.; Orange, J.S. Practical NK cell phenotyping and variability in healthy adults. Immunol. Res. 2015, 62, 341–356. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Tosato, F.; Bucciol, G.; Pantano, G.; Putti, M.C.; Sanzari, M.C.; Basso, G.; Plebani, M. Lymphocytes subsets reference values in childhood. Cytom. Part A 2015, 87, 81–85. [Google Scholar] [CrossRef]
- Tamura, A.; Inoue, S.; Mori, T.; Noguchi, J.; Nakamura, S.; Saito, A.; Kozaki, A.; Ishida, T.; Sadaoka, K.; Hasegawa, D.; et al. Low Multiplication Value of Absolute Monocyte Count and Absolute Lymphocyte Count at Diagnosis May Predict Poor Prognosis in Neuroblastoma. Front. Oncol. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Chung, H.S.; Higgins, G.R.; Siegel, S.E.; Seeger, R.C. Abnormalities of the immune system in children with neuroblastoma related to the neoplasm and chemotherapy. J. Pediatr. 1977, 90, 548–554. [Google Scholar] [CrossRef]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC class I downregulation in cancer: Underlying mechanisms and potential targets for cancer immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Domingos-pereira, S.; Galliverti, G.; Hanahan, D.; Nardelli-haefliger, D. Intravaginal CpG as tri-therapy towards efficient regression of genital HPV16 tumors. J. Immunol. Ther. Cancer 2019, 1, 1–7. [Google Scholar]
- Chitadze, G.; Lettau, M.; Luecke, S.; Wang, T.; Janssen, O.; Fürst, D.; Mytilineos, J.; Wesch, D.; Oberg, H.H.; Held-Feindt, J.; et al. NKG2D- and T-cell receptor-dependent lysis of malignant glioma cell lines by human γδ T cells: Modulation by temozolomide and A disintegrin and metalloproteases 10 and 17 inhibitors. Oncoimmunology 2016, 5, 1–13. [Google Scholar] [CrossRef]
- Rosanda, C.; De Bernardi, B.; Pasino, M.; Bisogni, M.C.; Maggio, A.; Haupt, R.; Tonini, G.P.; Ponzoni, M. Immune Evaluation of 50 Children with Neuroblastoma at Onset. Med. Pediatr. Oncol. 1982. [Google Scholar] [CrossRef]
- Helson, L.; Shou, L.; Tauber, J. Lymphocyte transformation in children with neuroblastoma. J. Natl. Cancer Inst. 1976, 57, 721–722. [Google Scholar] [CrossRef]
- Pelizzo, G.; Veschi, V.; Mantelli, M.; Croce, S.; Di Benedetto, V.; D’Angelo, P.; Maltese, A.; Catenacci, L.; Apuzzo, T.; Scavo, E.; et al. Microenvironment in neuroblastoma: Isolation and characterization of tumor-derived mesenchymal stromal cells. BMC Cancer 2018, 18, 1176. [Google Scholar] [CrossRef] [Green Version]
- Mussai, F.; Egan, S.; Hunter, S.; Webber, H.; Fisher, J.; Wheat, R.; McConville, C.; Sbirkov, Y.; Wheeler, K.; Bendle, G.; et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res. 2015, 75, 3043–3053. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ding, G.; Xu, X. Immunomodulatory functions of mesenchymal stem cells and possible mechanisms. Histol. Histopathol. 2016, 31, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zea, A.H.; Rodriguez, P.C.; Culotta, K.S.; Hernandez, C.P.; DeSalvo, J.; Ochoa, J.B.; Park, H.J.; Zabaleta, J.; Ochoa, A.C. L-Arginine modulates CD3ζ expression and T cell function in activated human T lymphocytes. Cell. Immunol. 2004, 232, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Troschke-Meurer, S.; Siebert, N.; Marx, M.; Zumpe, M.; Ehlert, K.; Mutschlechner, O.; Loibner, H.; Ladenstein, R.; Lode, H.N. Low CD4+/CD25+/CD127− regulatory T cell- and high INF-γ levels are associated with improved survival of neuroblastoma patients treated with long-term infusion of ch14.18/CHO combined with interleukin-2. Oncoimmunology 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Gershan, J.A.; Johnson, B.D. Depletion of CD4 T cells enhances immunotherapy for neuroblastoma after syngeneic HSCT but compromises development of antitumor immune memory. Blood 2009, 113, 4449–4457. [Google Scholar] [CrossRef] [Green Version]
- Jing, W.; Yan, X.; Hallett, W.H.D.; Gershan, J.A.; Johnson, B.D. Depletion of CD25+ T cells from hematopoietic stem cell grafts increases posttransplantation vaccine-induced immunity to neuroblastoma. Blood 2011. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.D.; Jing, W.; Orentas, R.J. CD25+ regulatory T cell inhibition enhances vaccine-induced immunity to neuroblastoma. J. Immunother. 2007, 30, 203–214. [Google Scholar] [CrossRef]
- Charych, D.H.; Hoch, U.; Langowski, J.L.; Lee, S.R.; Addepalli, M.K.; Kirk, P.B.; Sheng, D.; Liu, X.; Sims, P.W.; VanderVeen, L.A.; et al. NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clin. Cancer Res. 2016, 22, 680–690. [Google Scholar] [CrossRef] [Green Version]
- Diab, A.; Tannir, N.M.; Bentebibel, S.E.; Hwu, P.; Papadimitrakopoulou, V.; Haymaker, C.; Kluger, H.M.; Gettinger, S.N.; Sznol, M.; Tykodi, S.S.; et al. Bempegaldesleukin (NKTR-214) plus Nivolumab in Patients with Advanced Solid Tumors: Phase I Dose-Escalation Study of Safety, Efficacy, and Immune Activation (PIVOT-02). Cancer Discov. 2020, 10, 1158–1173. [Google Scholar] [CrossRef]
- Waldmann, T.A. The biology of interleukin-2 and interleukin-15: Implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006, 6, 595–601. [Google Scholar] [CrossRef]
- Heinze, A.; Grebe, B.; Bremm, M.; Huenecke, S.; Munir, T.A.; Graafen, L.; Frueh, J.T.; Merker, M.; Rettinger, E.; Soerensen, J.; et al. The Synergistic Use of IL-15 and IL-21 for the Generation of NK Cells From CD3/CD19-Depleted Grafts Improves Their ex vivo Expansion and Cytotoxic Potential against Neuroblastoma: Perspective for Optimized Immunotherapy Post Haploidentical Stem Cell Trans. Front. Immunol. 2019, 10, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Modak, S.; Le Luduec, J.B.; Cheung, I.Y.; Goldman, D.A.; Ostrovnaya, I.; Doubrovina, E.; Basu, E.; Kushner, B.H.; Kramer, K.; Roberts, S.S.; et al. Adoptive immunotherapy with haploidentical natural killer cells and Anti-GD2 monoclonal antibody m3F8 for resistant neuroblastoma: Results of a phase I study. Oncoimmunology 2018, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | Total (n = 25) |
|---|---|
| Gender male female | 14 (56%) 11 (44%) |
| Median age at diagnosis, year, (range) | 3.9 (0.3–10.8) |
| Stage 3 disease | 1 (4%) |
| Stage 4 disease | 24 (96%) |
| Genetics | |
| MYCN Neg Gain Amp | 18 (72%) 2 (8%) 5 (20%) |
| 1p normal partial loss loss gain | 14 (56%) 9 (36%) 1 (4%) 1 (4%) |
| 17q normal partial gain gain unknown | 1 (4%) 10 (40%) 11 (44%) 3 (12%) |
| ALK mutation Yes no gain unknown | 5 (20%) 16 (64%) 1 (4%) 3 (12%) |
| CR or PR after induction chemotherapy (3× N5/N6) | 14 (56%) |
| HD + ASCT | 20 (80%) |
| Conditioning Regimen | |
| Busulfan/melphalan | 14/20 (70%) |
| Carboplatin/etoposide/melphalan | 6/20 (30%) |
| CD34+ cell dose ×106/kg, (range) | 2.47 (0.59–21.73) |
| Immunotherapy | 16 (64%) |
| Time to immunotherapy, d, (range) | 137 (108–193) |
| Event: progression or relapse | 7 (31%) |
| Event: Refractory Disease | 3 (14%) |
| Event: Toxicity | 1 (5%) |
| Alive at last FU | 14 (56%) |
| Median EFS, year (range) | 1.65 (0.11–3.67) |
| Median follow-up OS, year, (range) | 2.14 (0.65–3.67) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szanto, C.L.; Cornel, A.M.; Tamminga, S.M.; Delemarre, E.M.; de Koning, C.C.H.; van den Beemt, D.A.M.H.; Dunnebach, E.; Tas, M.L.; Dierselhuis, M.P.; Tytgat, L.G.A.M.; et al. Immune Monitoring during Therapy Reveals Activitory and Regulatory Immune Responses in High-Risk Neuroblastoma. Cancers 2021, 13, 2096. https://doi.org/10.3390/cancers13092096
Szanto CL, Cornel AM, Tamminga SM, Delemarre EM, de Koning CCH, van den Beemt DAMH, Dunnebach E, Tas ML, Dierselhuis MP, Tytgat LGAM, et al. Immune Monitoring during Therapy Reveals Activitory and Regulatory Immune Responses in High-Risk Neuroblastoma. Cancers. 2021; 13(9):2096. https://doi.org/10.3390/cancers13092096
Chicago/Turabian StyleSzanto, Celina L., Annelisa M. Cornel, Sara M. Tamminga, Eveline M. Delemarre, Coco C. H. de Koning, Denise A. M. H. van den Beemt, Ester Dunnebach, Michelle L. Tas, Miranda P. Dierselhuis, Lieve G. A. M. Tytgat, and et al. 2021. "Immune Monitoring during Therapy Reveals Activitory and Regulatory Immune Responses in High-Risk Neuroblastoma" Cancers 13, no. 9: 2096. https://doi.org/10.3390/cancers13092096
APA StyleSzanto, C. L., Cornel, A. M., Tamminga, S. M., Delemarre, E. M., de Koning, C. C. H., van den Beemt, D. A. M. H., Dunnebach, E., Tas, M. L., Dierselhuis, M. P., Tytgat, L. G. A. M., van Noesel, M. M., Kraal, K. C. J. M., Boelens, J. -J., Huitema, A. D. R., & Nierkens, S. (2021). Immune Monitoring during Therapy Reveals Activitory and Regulatory Immune Responses in High-Risk Neuroblastoma. Cancers, 13(9), 2096. https://doi.org/10.3390/cancers13092096