
The Role of Senescent Cells in Acquired Drug Resistance and Secondary Cancer in BRAFi-Treated Melanoma

Abstract
:Simple Summary
Abstract
1. Introduction
2. BRAF Oncogene, Common Mutation BRAFV600E
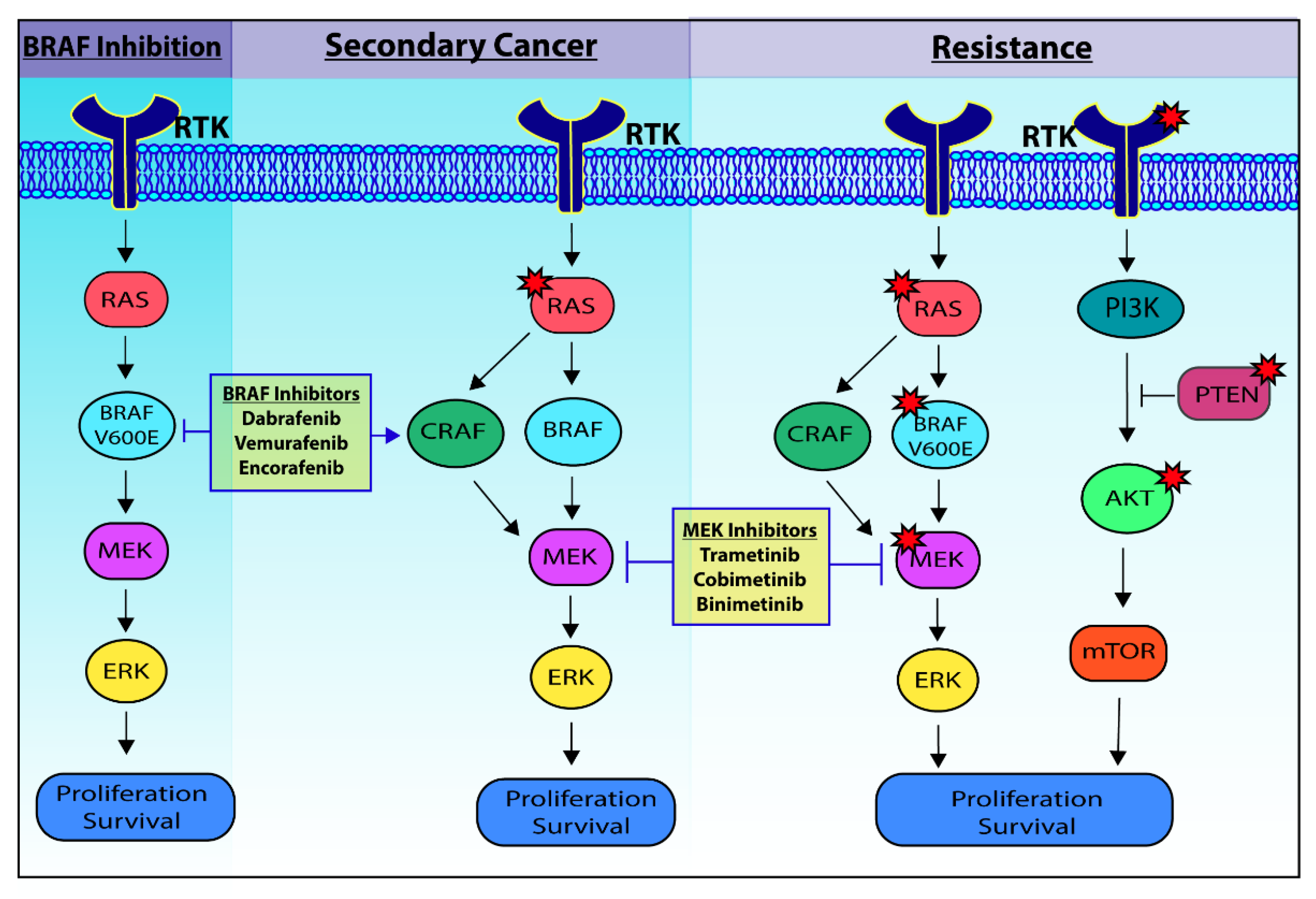
3. BRAF Inhibitors, Resistance and Secondary Cancer
4. Addressing Secondary Cancer and Resistance to BRAF Inhibitors
5. Senescence in Tumorigenesis and Melanoma
6. Future Therapy Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts & Figures 2020. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2020.html (accessed on 16 April 2021).
- National Cancer Institute. Melanoma of the Skin—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/melan.html (accessed on 16 April 2021).
- Miller, A.J.; Mihm, M.C. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Berwick, M.; Erdei, E.; Hay, J. Melanoma Epidemiology and Public Health. Dermatol. Clin. 2009, 27, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, G.C.; Candido, S.; Falzone, L.; Spandidos, D.A.; Libra, M. Cutaneous melanoma and the immunotherapy revolution (Review). Int. J. Oncol. 2020, 57, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Yuan, L.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [Green Version]
- Mehnert, J.M.; Kluger, H.M. Driver Mutations in Melanoma: Lessons Learned From Bench-to-Bedside Studies. Curr. Oncol. Rep. 2012, 14, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Zhang, Z.; Liu, J.; Li, L.; Xu, X.; Yao, X.; Dai, Z.; Wang, X.; Yang, S.; Wu, H.; et al. Characterizations of Gene Alterations in Melanoma Patients from Chinese Population. BioMed Res. Int. 2020, 2020, 6096814. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Burd, C.E.; Liu, W.; Huynh, M.V.; Waqas, M.A.; Gillahan, J.E.; Clark, K.S.; Fu, K.; Martin, B.L.; Jeck, W.R.; Souroullas, G.P.; et al. Mutation-Specific RAS Oncogenicity Explains NRAS Codon 61 Selection in Melanoma. Cancer Discov. 2014, 4, 1418–1429. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sheikh, M.S. Melanoma: Molecular Pathogenesis and Therapeutic Management. Mol. Cell. Pharmacol. 2014, 6, 228. [Google Scholar]
- Chin, L. The genetics of malignant melanoma: Lessons from mouse and man. Nat. Rev. Cancer 2003, 3, 559–570. [Google Scholar] [CrossRef]
- Yang, G.; Rajadurai, A.; Tsao, H. Recurrent Patterns of Dual RB and p53 Pathway Inactivation in Melanoma. J. Investig. Dermatol. 2005, 125, 1242–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Hodi, F.S.; Fisher, D.E. From genes to drugs: Targeted strategies for melanoma. Nat. Rev. Cancer 2012, 12, 349–361. [Google Scholar] [CrossRef]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Carvajal, R.; Antonescu, C.; Wolchok, J.; Chapman, P.; Roman, R.; Teitcher, J.; Panageas, K.; Busam, K.; Chmielowski, B.; Lutzky, J.; et al. KIT as a Therapeutic Target in Metastatic Melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, A.J.-S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef] [PubMed]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.-I.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a–Rb pathway cooperate to enforce irreversible cellular senescence. Nature 2006, 8, 1291–1297. [Google Scholar] [CrossRef]
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage—how and why we age? eLife 2021, 10. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.J.; Oh, Y.S.; Park, S.C. Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. 2007, 14, 1020–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Kang, J.; Xia, J.; Li, Y.; Yang, B.; Chen, B.; Sun, W.; Song, X.; Xiang, W.; Wang, X.; et al. p53-related apoptosis resistance and tumor suppression activity in UVB-induced premature senescent human skin fibroblasts. Int. J. Mol. Med. 2008, 21, 645–653. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.-M.; DeMaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.N.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Hugdahl, E.; Kalvenes, M.B.; Puntervoll, H.E.; Ladstein, R.G.; A Akslen, L. BRAF-V600E expression in primary nodular melanoma is associated with aggressive tumour features and reduced survival. Br. J. Cancer 2016, 114, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and Clinicopathologic Associations of Oncogenic BRAF in Metastatic Melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.S.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.W.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Bennett, D.C. Human melanocyte senescence and melanoma susceptibility genes. Oncogene 2003, 22, 3063–3069. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Vredeveld, L.C.; Possik, P.A.; Smit, M.A.; Meissl, K.; Michaloglou, C.; Horlings, H.M.; Ajouaou, A.; Kortman, P.C.; Dankort, D.; McMahon, M.; et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev. 2012, 26, 1055–1069. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, C.; Kim, K.-H.; Sung, H.; Paraiso, K.H.T.; Dannenberg, J.-H.; Bosenberg, M.; Chin, L.; Kim, M. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene 2010, 29, 6222–6232. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment. Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E. UV Signature Mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, N.E.; Alexander, A.; Edmiston, S.N.; Parrish, E.; Millikan, R.C.; Berwick, M.; Groben, P.; Ollila, D.W.; Mattingly, D.; Conway, K. Tandem BRAF Mutations in Primary Invasive Melanomas. J. Investig. Dermatol. 2004, 122, 1245–1250. [Google Scholar] [CrossRef] [Green Version]
- Thomas, N.E.; Berwick, M.; Cordeiro-Stone, M. Could BRAF Mutations in Melanocytic Lesions Arise from DNA Damage Induced by Ultraviolet Radiation? J. Investig. Dermatol. 2006, 126, 1693–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastogi, R.P.; Richa; Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular Mechanisms of Ultraviolet Radiation-Induced DNA Damage and Repair. J. Nucleic Acids 2010, 2010, 592980. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Kong, Y.; Si, L.; Cui, C.; Sheng, X.; Chi, Z.; Dai, J.; Yu, S.; Ma, M.; Wu, X.; et al. Clinical significance of BRAFV600E mutation in circulating tumor DNA in Chinese patients with melanoma. Oncol. Lett. 2017, 15, 1839–1844. [Google Scholar] [CrossRef]
- Salemi, R.; Falzone, L.; Madonna, G.; Polesel, J.; Cinà, D.; Mallardo, D.; Ascierto, P.A.; Libra, M.; Candido, S. MMP-9 as a Candidate Marker of Response to BRAF Inhibitors in Melanoma Patients With BRAFV600E Mutation Detected in Circulating-Free DNA. Front. Pharmacol. 2018, 9, 856. [Google Scholar] [CrossRef]
- Ashida, A.; Sakaizawa, K.; Mikoshiba, A.; Uhara, H.; Okuyama, R. Quantitative analysis of the BRAF V600E mutation in circulating tumor-derived DNA in melanoma patients using competitive allele-specific TaqMan PCR. Int. J. Clin. Oncol. 2016, 21, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Jr, W.E.D.; You, M.J.; Depinho, R.A.; McMahon, M.; Bosenberg, M. BrafV600E cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Dhomen, N.; Reis-Filho, J.S.; Dias, S.D.R.; Hayward, R.; Savage, K.; Delmas, V.; LaRue, L.; Pritchard, C.; Marais, R. Oncogenic Braf Induces Melanocyte Senescence and Melanoma in Mice. Cancer Cell 2009, 15, 294–303. [Google Scholar] [CrossRef]
- Wellbrock, C.; Ogilvie, L.; Hedley, D.; Karasarides, M.; Martin, J.; Niculescu-Duvaz, D.; Springer, C.J.; Marais, R. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004, 64, 2338–2342. [Google Scholar] [CrossRef] [Green Version]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Krayem, M.; Najem, A.; Journe, F.; Morandini, R.; Sales, F.; Awada, A.; Ghanem, G.E. Acquired resistance to BRAFi reverses senescence-like phenotype in mutant BRAF melanoma. Oncotarget 2018, 9, 31888–31903. [Google Scholar] [CrossRef]
- Clynick, B.; Tabone, T.; Fuller, K.; Erber, W.; Meehan, K.; Millward, M.; Wood, B.A.; Harvey, N.T.; Fuller, K. Mutational Analysis of BRAF Inhibitor–Associated Squamoproliferative Lesions. J. Mol. Diagn. 2015, 17, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Flaherty, K.T. Resistance to BRAF-targeted therapy in melanoma. Eur. J. Cancer 2013, 49, 1297–1304. [Google Scholar] [CrossRef]
- Devji, T.; Levine, O.; Neupane, B.; Beyene, J.; Xie, F. Systemic Therapy for Previously Untreated Advanced BRAF-Mutated Melanoma. JAMA Oncol. 2017, 3, 366–373. [Google Scholar] [CrossRef]
- Nathanson, K.L.; Martin, A.-M.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.-T.; et al. Tumor Genetic Analyses of Patients with Metastatic Melanoma Treated with the BRAF Inhibitor Dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 19, 4868–4878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraiso, K.H.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.; et al. PTEN Loss Confers BRAF Inhibitor Resistance to Melanoma Cells through the Suppression of BIM Expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.; Niemetz-Rahn, A.; Nonnenmacher, A.; Tucholski, J.; Keilholz, U.; Fusi, A. Expression and activity of EGFR in human cutaneous melanoma cell lines and influence of vemurafenib on the EGFR pathway. Target. Oncol. 2014, 10, 77–84. [Google Scholar] [CrossRef]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF Receptor or SRC Family Kinase Signaling Overcomes BRAF Inhibitor Resistance in Melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, N.; Ma, X.; Bernard, D. Regulation of cellular senescence by retinoid X receptors and their partners. Mech. Ageing Dev. 2019, 183, 111131. [Google Scholar] [CrossRef]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment. Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef]
- Diazzi, S.; Tartare-Deckert, S.; Deckert, M. Bad Neighborhood: Fibrotic Stroma as a New Player in Melanoma Resistance to Targeted Therapies. Cancers 2020, 12, 1364. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, S.; Cheli, Y.; Ohanna, M.; Bonet, C.; Beuret, L.; Bille, K.; Loubat, A.; Hofman, V.; Hofman, P.; Ponzio, G.; et al. Microphthalmia-Associated Transcription Factor Controls the DNA Damage Response and a Lineage-Specific Senescence Program in Melanomas. Cancer Res. 2010, 70, 3813–3822. [Google Scholar] [CrossRef] [Green Version]
- Strub, T.; Giuliano, S.; Ye, T.; Bonet, C.; Keime, C.; Kobi, D.; Le Gras, S.; Cormont, M.; Ballotti, R.; Bertolotto, C.; et al. Essential role of microphthalmia transcription factor for DNA replication, mitosis and genomic stability in melanoma. Oncogene 2011, 30, 2319–2332. [Google Scholar] [CrossRef] [Green Version]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, K.; Suzuki, H.I. TGF-β Signaling in Cellular Senescence and Aging-Related Pathology. Int. J. Mol. Sci. 2019, 20, 5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.J.; Zhang, X.; Kumar, A.; Atkinson, E.J.; Zhu, Y.; Jachim, S.; Mazula, D.L.; Brown, A.K.; Berning, M.; Aversa, Z.; et al. The senescence-associated secretome as an indicator of age and medical risk. JCI Insight 2020, 5, 5. [Google Scholar] [CrossRef]
- Li, Q.; Li, J.; Wen, T.; Zeng, W.; Peng, C.; Yan, S.; Tan, J.; Yang, K.; Liu, S.; Guo, A.; et al. Overexpression of HMGB1 in melanoma predicts patient survival and suppression of HMGB1 induces cell cycle arrest and senescence in association with p21 (Waf1/Cip1) up-regulation via a p53-independent, Sp1-dependent pathway. Oncotarget 2014, 5, 6387–6403. [Google Scholar] [CrossRef] [Green Version]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar] [CrossRef]
- Thies, A.; Moll, I.; Berger, J.; Wagener, C.; Brümmer, J.; Schulze, H.-J.; Brunner, G.; Schumacher, U. CEACAM1 Expression in Cutaneous Malignant Melanoma Predicts the Development of Metastatic Disease. J. Clin. Oncol. 2002, 20, 2530–2536. [Google Scholar] [CrossRef]
- Wicklein, D.; Otto, B.; Suling, A.; Elies, E.; Lüers, G.; Lange, T.; Feldhaus, S.; Maar, H.; Schröder-Schwarz, J.; Brunner, G.; et al. CEACAM1 promotes melanoma metastasis and is involved in the regulation of the EMT associated gene network in melanoma cells. Sci. Rep. 2018, 8, 11893. [Google Scholar] [CrossRef] [Green Version]
- Helfrich, I.; Singer, B.B. Size Matters: The Functional Role of the CEACAM1 Isoform Signature and Its Impact for NK Cell-Mediated Killing in Melanoma. Cancers 2019, 11, 356. [Google Scholar] [CrossRef] [Green Version]
- Sappino, A.-P.; Buser, R.; Seguin, Q.; Fernet, M.; Lesne, L.; Gumypause, F.; Reith, W.; Favaudon, V.; Mandriota, S.J. The CEACAM1 tumor suppressor is an ATM and p53-regulated gene required for the induction of cellular senescence by DNA damage. Oncogenesis 2012, 1, e7. [Google Scholar] [CrossRef] [Green Version]
- Kfir-Elirachman, K.; Ortenberg, R.; Vizel, B.; Besser, M.J.; Barshack, I.; Schachter, J.; Nemlich, Y.; Markel, G. Regulation of CEACAM1 Protein Expression by the Transcription Factor ETS-1 in BRAF-Mutant Human Metastatic Melanoma Cells. Neoplasia 2018, 20, 401–409. [Google Scholar] [CrossRef]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 Expression Is Associated With High Proliferation Rate and Aggressive Tumor Subgroups in Cutaneous Melanoma and Cancers of the Endometrium, Prostate, and Breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Fan, T.; Jiang, S.; Chung, N.; Alikhan, A.; Ni, C.; Lee, C.-C.R.; Hornyak, T.J. EZH2-Dependent Suppression of a Cellular Senescence Phenotype in Melanoma Cells by Inhibition of p21/CDKN1A Expression. Mol. Cancer Res. 2011, 9, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin Remodeling Underlies the Senescence-Associated Secretory Phenotype of Tumor Stromal Fibroblasts That Supports Cancer Progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.C.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RASMutations in Cutaneous Squamous-Cell Carcinomas in Patients Treated with BRAF Inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doma, E.; Rupp, C.; Varga, A.; Kern, F.; Riegler, B.; Baccarini, M. Skin Tumorigenesis Stimulated by Raf Inhibitors Relies Upon Raf Functions That Are Dependent and Independent of ERK. Cancer Res. 2013, 73, 6926–6937. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Klimek, V.M.; Gaskell, A.A.; Viale, A.; Cheng, D.; Kim, E.; Rampal, R.; Bluth, M.; Harding, J.J.; Callahan, M.K.; et al. Efficacy of Intermittent Combined RAF and MEK Inhibition in a Patient with Concurrent BRAF- and NRAS-Mutant Malignancies. Cancer Discov. 2014, 4, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Callahan, M.K.; Rampal, R.; Harding, J.J.; Klimek, V.M.; Chung, Y.R.; Merghoub, T.; Wolchok, J.D.; Solit, D.B.; Rosen, N.; Abdel-Wahab, O.; et al. Progression of RAS-Mutant Leukemia during RAF Inhibitor Treatment. N. Engl. J. Med. 2012, 367, 2316–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, M.C.; Behren, A.; Chionh, F.; Mariadason, J.; Vella, L.J.; Do, H.; Dobrovic, A.; Tebbutt, N.; Cebon, J. BRAF Inhibitor–Driven Tumor Proliferation in a KRAS-Mutated Colon Carcinoma Is Not Overcome by MEK1/2 Inhibition. J. Clin. Oncol. 2013, 31, e448–e451. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cohen, D.; Rady, P.; Tyring, S. BRAF inhibitor-associated cutaneous squamous cell carcinoma: New mechanistic insight, emerging evidence for viral involvement and perspectives on clinical management. Br. J. Dermatol. 2017, 177, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Purdie, K.J.; Proby, C.M.; Rizvi, H.; Griffin, H.; Doorbar, J.; Sommerlad, M.; Feltkamp, M.C.; Van Der Meijden, E.; Inman, G.J.; South, A.P.; et al. The Role of Human Papillomaviruses and Polyomaviruses in BRAF-Inhibitor Induced Cutaneous Squamous Cell Carcinoma and Benign Squamoproliferative Lesions. Front. Microbiol. 2018, 9, 1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, D.N.; Lawson, S.K.; Shaver, A.C.; Du, L.; Nguyen, H.P.; He, Q.; Johnson, D.B.; Lumbang, W.A.; Moody, B.R.; Prescott, J.L.; et al. Contribution of Beta-HPV Infection and UV Damage to Rapid-Onset Cutaneous Squamous Cell Carcinoma during BRAF-Inhibition Therapy. Clin. Cancer Res. 2015, 21, 2624–2634. [Google Scholar] [CrossRef] [Green Version]
- Paraiso, K.H.T.; Fedorenko, I.V.; Cantini, L.P.; Munko, A.C.; Hall, M.; Sondak, V.K.; Messina, J.L.; Flaherty, K.T.; Smalley, K.S.M. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 2010, 102, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Corrie, P.G. Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Ther. Adv. Med Oncol. 2015, 7, 122–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. CANCER: Suppressing Cancer: The Importance of Being Senescent. Science 2005, 309, 886–887. [Google Scholar] [CrossRef] [Green Version]
- DeMaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resnik, S.R.; Egger, A.; Abujamra, B.A.; Jozic, I. Clinical Implications of Cellular Senescence on Wound Healing. Curr. Dermatol. Rep. 2020, 9, 286–297. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguría, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef]
- Vernot, J.P. Senescence-Associated Pro-inflammatory Cytokines and Tumor Cell Plasticity. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Milanovic, M.; Yu, Y.; Schmitt, C.A. The Senescence–Stemness Alliance—A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018, 28, 1049–1061. [Google Scholar] [CrossRef]
- von Zglinicki, T.; Saretzki, G.; Ladhoff, J.; di Fagagna, F.D.; Jackson, S. Human cell senescence as a DNA damage response. Mech. Ageing Dev. 2005, 126, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.M.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.L.D.; Gackowska, A.; Anderson, R.; Taschuk, M.T.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial Dysfunction Accounts for the Stochastic Heterogeneity in Telomere-Dependent Senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef]
- Korkaya, H.; Liu, S.; Wicha, M.S. Regulation of Cancer Stem Cells by Cytokine Networks: Attacking Cancer’s Inflammatory Roots: Figure 1. Clin. Cancer Res. 2011, 17, 6125–6129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laberge, R.-M.; Awad, P.; Campisi, J.; Desprez, P.-Y. Epithelial-Mesenchymal Transition Induced by Senescent Fibroblasts. Cancer Microenviron. 2011, 5, 39–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef] [Green Version]
- Prieto, L.I.; Baker, D.J. Cellular Senescence and the Immune System in Cancer. Gerontology 2019, 65, 505–512. [Google Scholar] [CrossRef]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A key player in cancer development. J. Hematol. Oncol. 2020, 13, 1–18. [Google Scholar] [CrossRef]
- Su, D.-M.; Aw, D.; Palmer, D.B. Immunosenescence: A product of the environment? Curr. Opin. Immunol. 2013, 25, 498–503. [Google Scholar] [CrossRef]
- Battram, A.M.; Bachiller, M.; Martín-Antonio, B. Senescence in the Development and Response to Cancer with Immunotherapy: A Double-Edged Sword. Int. J. Mol. Sci. 2020, 21, 4346. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.C.; Cheong, S.C.; Chong, H.; Chow, J.; Moss, T.; Abdel-Malek, Z.A.; Marais, R.; Wynford-Thomas, D.; Bennett, D.C. Cellular senescence in naevi and immortalisation in melanoma: A role for p16? Br. J. Cancer 2006, 95, 496–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suram, A.; Kaplunov, J.; Patel, P.L.; Ruan, H.; Cerutti, A.; Boccardi, V.; Fumagalli, M.; Di Micco, R.; Mirani, N.; Gurung, R.L.; et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012, 31, 2839–2851. [Google Scholar] [CrossRef]
- Boussemart, L.; Routier, E.; Mateus, C.; Opletalova, K.; Sebille, G.; Kamsu-Kom, N.; Thomas, M.; Vagner, S.; Favre, M.; Tomasic, G.; et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: A study of 42 patients. Ann. Oncol. 2013, 24, 1691–1697. [Google Scholar] [CrossRef]
- Anforth, R.; Blumetti, T.; Kefford, R.; Sharma, R.; Scolyer, R.; Kossard, S.; Long, G.; Fernandez-Peñas, P. Cutaneous manifestations of dabrafenib (GSK2118436): A selective inhibitor of mutant BRAF in patients with metastatic melanoma. Br. J. Dermatol. 2012, 167, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.I.; Francis, D.A.; Karpova, A.Y.; Dowhanick, J.J.; Benson, J.D.; Howley, P.M. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21CIP-dependent pathways. EMBO J. 2000, 19, 5762–5771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellmann, L.; Cappellano, G.; Schachtl-Riess, J.F.; Prokopi, A.; Seretis, A.; Ortner, D.; Tripp, C.H.; Brinckerhoff, C.E.; Mullins, D.W.; Stoitzner, P. A TLR7 agonist strengthens T and NK cell function during BRAF-targeted therapy in a preclinical melanoma model. Int. J. Cancer 2019, 146, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Xue, G. Major Physiological Signaling Pathways in the Regulation of Cell Proliferation and Survival. Handb. Exp. Pharmacol. 2018, 249, 13–30. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.E.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Mavrogonatou, E.; Pratsinis, H.; Kletsas, D. The role of senescence in cancer development. Semin. Cancer Biol. 2020, 62, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedernhofer, L.J.; Robbins, P.D. Senotherapeutics for healthy ageing. Nat. Rev. Drug Discov. 2018, 17, 377. [Google Scholar] [CrossRef] [Green Version]
- Pal, H.C.; Sharma, S.; Strickland, L.R.; Katiyar, S.K.; Ballestas, M.E.; Athar, M.; Elmets, C.A.; Afaq, F. Fisetin Inhibits Human Melanoma Cell Invasion through Promotion of Mesenchymal to Epithelial Transition and by Targeting MAPK and NFκB Signaling Pathways. PLoS ONE 2014, 9, e86338. [Google Scholar] [CrossRef] [Green Version]
- Syed, D.; Adhami, V.; Khan, M.; Mukhtar, H. Inhibition of Akt/mTOR Signaling by the Dietary Flavonoid Fisetin. Anti-Cancer Agents Med. Chem. 2013, 13, 995–1001. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Esnault, S.; Adhami, V.M.; Noll, A.L.; Banang-Mbeumi, S.; Roy, T.; Singh, S.S.; Huang, S.; Kousoulas, K.G.; Mukhtar, H. Fisetin, a 3,7,3’,4’-Tetrahydroxyflavone Inhibits the PI3K/Akt/mTOR and MAPK Pathways and Ameliorates Psoriasis Pathology in 2D and 3D Organotypic Human Inflammatory Skin Models. Cells 2019, 8, 1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caltagirone, S.; Rossi, C.; Poggi, A.; Ranelletti, F.O.; Natali, P.G.; Brunetti, M.; Aiello, F.B.; Piantelli, M. Flavonoids apigenin and quercetin inhibit melanoma growth and metastatic potential. Int. J. Cancer 2000, 87, 595–600. [Google Scholar] [CrossRef]
- Harris, Z.; Donovan, M.G.; Branco, G.M.; Limesand, K.H.; Burd, R. Quercetin as an Emerging Anti-Melanoma Agent: A Four-Focus Area Therapeutic Development Strategy. Front. Nutr. 2016, 3. [Google Scholar] [CrossRef]
- Thangasamy, T.; Sittadjody, S.; Lanza-Jacoby, S.; Wachsberger, P.R.; Limesand, K.H.; Burd, R. Quercetin Selectively Inhibits Bioreduction and Enhances Apoptosis in Melanoma Cells That Overexpress Tyrosinase. Nutr. Cancer 2007, 59, 258–268. [Google Scholar] [CrossRef]
- Song, X.; Gao, T.; Lei, Q.; Zhang, L.; Yao, Y.; Xiong, J. Piperlongumine Induces Apoptosis in Human Melanoma Cells Via Reactive Oxygen Species Mediated Mitochondria Disruption. Nutr. Cancer 2018, 70, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; van Deursen, J.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anti-Cancer Therapy. J. Natl. Cancer Inst. 2021. [Google Scholar] [CrossRef]
- Pacey, S.; Wilson, R.H.; Walton, M.; Eatock, M.M.; Hardcastle, A.; Zetterlund, A.; Arkenau, H.-T.; Moreno-Farre, J.; Banerji, U.; Roels, B.; et al. A Phase I Study of the Heat Shock Protein 90 Inhibitor Alvespimycin (17-DMAG) Given Intravenously to Patients with Advanced Solid Tumors. Clin. Cancer Res. 2011, 17, 1561–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.K.; Jeong, K.-H.; Choi, H.; Ahn, H.-J.; Lee, M.-H. Heat shock protein 90 inhibitor enhances apoptosis by inhibiting the AKT pathway in thermal-stimulated SK-MEL-2 human melanoma cell line. J. Dermatol. Sci. 2018, 90, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pacey, S.; Gore, M.; Chao, D.; Banerji, U.; Larkin, J.; Sarker, S.; Owen, K.; Asad, Y.; Raynaud, F.; Walton, M.; et al. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Investig. New Drugs 2010, 30, 341–349. [Google Scholar] [CrossRef]
- Solit, D.B.; Osman, I.; Polsky, D.; Panageas, K.S.; Daud, A.; Goydos, J.S.; Teitcher, J.; Wolchok, J.D.; Germino, F.J.; Krown, S.E.; et al. Phase II Trial of 17-Allylamino-17-Demethoxygeldanamycin in Patients with Metastatic Melanoma. Clin. Cancer Res. 2008, 14, 8302–8307. [Google Scholar] [CrossRef] [Green Version]
- Vaishampayan, U.N.; Burger, A.M.; Sausville, E.A.; Heilbrun, L.K.; Li, J.; Horiba, M.N.; Egorin, M.J.; Ivy, P.; Pacey, S.; Lorusso, P.M. Safety, Efficacy, Pharmacokinetics, and Pharmacodynamics of the Combination of Sorafenib and Tanespimycin. Clin. Cancer Res. 2010, 16, 3795–3804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankel, A.E.; Eskiocak, U.; Gill, J.G.; Yuan, S.; Ramesh, V.; Froehlich, T.W.; Ahn, C.; Morrison, S.J. Digoxin Plus Trametinib Therapy Achieves Disease Control in BRAF Wild-Type Metastatic Melanoma Patients. Neoplasia 2017, 19, 255–260. [Google Scholar] [CrossRef]
- Sale, M.J.; Cook, S.J. The BH3 mimetic ABT-263 synergizes with the MEK1/2 inhibitor selumetinib/AZD6244 to promote BIM-dependent tumour cell death and inhibit acquired resistance. Biochem. J. 2013, 450, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Kalinsky, K.; Lee, S.; Np, K.M.R.; Lawrence, D.P.; Iafrarte, A.J.; Borger, D.R.; Margolin, K.A.; Leitao, M.M., Jr.; Tarhini, A.A.; Koon, H.B.; et al. A phase 2 trial of dasatinib in patients with locally advanced or stage IV mucosal, acral, or vulvovaginal melanoma: A trial of the ECOG-ACRIN Cancer Research Group (E2607). Cancer 2017, 123, 2688–2697. [Google Scholar] [CrossRef]
- Kluger, H.M.; Dudek, A.Z.; McCann, C.; Ritacco, J.; Southard, N.; Jilaveanu, L.B.; Molinaro, A.; Sznol, M. A phase 2 trial of dasatinib in advanced melanoma. Cancer 2011, 117, 2202–2208. [Google Scholar] [CrossRef]
- Algazi, A.P.; Weber, J.S.; Andrews, S.C.; Urbas, P.; Munster, P.N.; DeConti, R.C.; Hwang, J.; Sondak, V.K.; Messina, J.L.; McCalmont, T.; et al. Phase I clinical trial of the Src inhibitor dasatinib with dacarbazine in metastatic melanoma. Br. J. Cancer 2011, 106, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, C.; Leon-Ferre, R.; Laux, D.; Deutsch, J.; Smith, B.J.; Frees, M.; Milhem, M. Treatment of resistant metastatic melanoma using sequential epigenetic therapy (decitabine and panobinostat) combined with chemotherapy (temozolomide). Cancer Chemother. Pharmacol. 2014, 74, 691–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, N.; Buchbinder, E.I.; Granter, S.R.; Rodig, S.J.; Giobbie-Hurder, A.; Becerra, C.; Tsiaras, A.; Gjini, E.; Fisher, D.E.; Hodi, F.S. A phase I trial of panobinostat (LBH 589) in patients with metastatic melanoma. Cancer Med. 2016, 5, 3041–3050. [Google Scholar] [CrossRef]
- He, Y.; Junling, Z.; Zhang, J.; Yang, Y.; Qian, Y.-W.; Zhou, D. The curcumin analog EF24 is highly active against chemotherapy-resistant melanoma cells. Curr. Cancer Drug Targets 2021, 21, 1. [Google Scholar] [CrossRef]
- Da Silva, J.M.C.; Campos, M.L.A.; Teixeira, M.P.; Faustino, R.D.S.; Aleixo, R.C.; Cavalcante, F.J.P.; Gomes, L.R.O.; De Albuquerque, L.Z.; Azevedo, A.D.N.; Cabral, V.R.; et al. Ouabain pre-treatment modulates B and T lymphocytes and improves survival of melanoma-bearing animals. Int. Immunopharmacol. 2020, 86, 106772. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cai, W.; Han, B.; Zhang, J.; Yu, B.; Chen, M. Ouabain Exhibited Strong Anticancer Effects in Melanoma Cells via Induction of Apoptosis, G2/M Phase Arrest, and Migration Inhibition. OncoTargets Ther. 2021, 14, 1261–1273. [Google Scholar] [CrossRef]
- Wroblewski, D.; Mijatov, B.; Mohana-Kumaran, N.; Lai, F.; Gallagher, S.J.; Haass, N.K.; Zhang, X.D.; Hersey, P. The BH3-mimetic ABT-737 sensitizes human melanoma cells to apoptosis induced by selective BRAF inhibitors but does not reverse acquired resistance. Carcinogenesis 2012, 34, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, N.; Almeida, A.; Partyka, K.A.; Lu, Y.; Schwan, J.V.; Lambert, K.; Rogers, M.; Robinson, W.A.; Robinson, S.E.; Applegate, A.J.; et al. Combining a GSI and BCL-2 inhibitor to overcome melanoma’s resistance to current treatments. Oncotarget 2016, 7, 84594–84607. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Cheng, X.; Zhou, X. The BET-bromodomain inhibitor JQ1 mitigates vemurafenib drug resistance in melanoma. Melanoma Res. 2018, 28, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Yoo, E.-S.; Woo, J.-S.; Han, S.-H.; Lee, J.-H.; Jung, S.-H.; Kim, H.-J.; Jung, J.-Y. Antitumor and apoptotic effects of quercetin on human melanoma cells involving JNK/P38 MAPK signaling activation. Eur. J. Pharmacol. 2019, 860, 172568. [Google Scholar] [CrossRef]
- Soll, F.; Ternent, C.; Berry, I.M.; Kumari, D.; Moore, T.C. Quercetin Inhibits Proliferation and Induces Apoptosis of B16 Melanoma Cells In Vitro. ASSAY Drug Dev. Technol. 2020, 18, 261–268. [Google Scholar] [CrossRef]
- Wang, K.; Hu, D.-N.; Lin, H.-W.; Yang, W.-E.; Hsieh, Y.-H.; Chien, H.-W.; Yang, S.-F. Fisetin induces apoptosis through mitochondrial apoptosis pathway in human uveal melanoma cells. Environ. Toxicol. 2018, 33, 527–534. [Google Scholar] [CrossRef]
- Sechi, M.; Lall, R.K.; Afolabi, S.O.; Singh, A.; Joshi, D.C.; Chiu, S.-Y.; Mukhtar, H.; Syed, D.N. Fisetin targets YB-1/RSK axis independent of its effect on ERK signaling: Insights from in vitro and in vivo melanoma models. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Liu-Smith, F.; Meyskens, F.L. Molecular mechanisms of flavonoids in melanin synthesis and the potential for the prevention and treatment of melanoma. Mol. Nutr. Food Res. 2016, 60, 1264–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Mechanisms of Action | Treatment | Developmental Stage |
|---|---|---|---|
| Alvespimycin | HSP inhibitor | Alvespimycin hydrochloride | Clinical Trail: Phase 1; NCT00089362; Metastatic or unresectable solid tumors including melanoma |
| Alvespimycin hydrochloride | Phase 1; NCT00248521; Adult solid tumor including melanoma [146] | ||
| Alvespimycin hydrochloride | Discovery Phase: human melanoma cell line [147] | ||
| Tanespimycin | HSP inhibitor | Tanespimycin | Clinical Trail: Phase 2; NCT00087386; Recurrent or phase III, IV melanoma |
| Tanespimycin | Phase 1; NCT00004065; Refractory advanced solid tumors including melanoma or hematologic cancer | ||
| Tanespimycin | Phase 2; NCT00104897; Metastatic malignant melanoma [148] | ||
| Tanespimycin | Phase 2; Metastatic Melanoma [149] | ||
| Tanespimycin and Sorafenib | Phase 1; Melanoma, renal cancer and colorectal cancer [150] | ||
| Digoxin | Na+/K+ ATPase inhibitor | Trametinib and Digoxin | Clinical Trail: Phase 1; NCT02138292; Unresectable or metastatic BRAF wild-type melanoma [151] |
| Vemurafenib and Digoxin | Phase 1; NCT01765569; Advanced BRAFV600 mutant melanoma | ||
| Navitoclax (ABT-263) | Bcl-2/Bcl-xL inhibitor | Dabrafenib, trametinib, and navitoclax | Clinical Trail: Phase 1/2; NCT01989585; BRAF mutant melanoma or unresectable or metastatic solid tumors |
| Novitoclax and selumetinib | Discovery Phase: Melanoma cell lines [152] | ||
| Dasatinib | Pan receptor tyrosine kinase inhibitor | Dasatinib | Clinical Trail: Phase 2; NCT00700882; Melanoma (Skin) [153] |
| Dendritic cell Vaccines + Dasatinib | Phase 2; NCT01876212; Metastatic melanoma | ||
| Dasatinib and Dacarbazine | Phase 1/2; NCT00597038; Metastatic Melanoma | ||
| Dasatinib | Phase 2; NCT00436605; Unresectable stage III melanoma or stage IV melanoma | ||
| Dasatinib | Phase 1; Advanced melanoma [154] | ||
| Dasatinib and Dacarbazine | Phase 1; Metastatic melanoma [155] | ||
| Dasatinib | Discovery Phase: Melanoma cell lines [67] | ||
| Panobinostat (LBH589) | Pan HDAC inhibitor | Panobinostat | Clinical Trail: Phase 1; NCT01065467; Metastatic Melanoma |
| Panobinostat and Ipilimumab | Phase 1; NCT02032810; Unresectable stage III/IV melanoma | ||
| Temozolomide, Decitabine, Panobinostat | Phase 1/2; NCT00925132; Metastatic Melanoma [156] | ||
| Panobinostat (LBH589) | Phase 1; metastatic melanoma [157] | ||
| Curcumin analog, EF24 | Promote degradation of anti-apoptotic Bcl-2 proteins | EF24 | Discovery Phase: Malignant melanoma cell lines [158] |
| Piperlongumine | Targets OXR1 | Piperlongumine | Discovery Phase: Human melanoma cell [143] |
| Ouabain | Na+/K+ ATPase inhibitor | Ouabain | Discovery Phase: Malignant melanoma cell lines [159,160] |
| ABT-737 | Bcl-2/Bcl-xL inhibitor | ABT-737 and PLX4720 | Discovery Phase: Human melanoma cell lines and primary melanoma cell culture [161] |
| ABT-737 and GSI (γ-Secretase Inhibitor) | Non-MIC (bulk of melanoma) and MICs [162] | ||
| JQ1 | BET inhibitor | JQ1 and vemurafenib | Discovery Phase: BRAF mutant vemurafenib-resistant melanoma cells [163] |
| Quercetin | Activates estrogen receptors and inhibits PI3 kinase | Quercetin | Discovery Phase: Melanoma cell lines [140,141,142,164,165] |
| Fisetin | Blocks PI3K/AKT/mTOR pathway | Fisetin | Discovery Phase: Melanoma cell lines [137,138,166,167,168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, E.L.; Hu, J.J.; Niedernhofer, L.J. The Role of Senescent Cells in Acquired Drug Resistance and Secondary Cancer in BRAFi-Treated Melanoma. Cancers 2021, 13, 2241. https://doi.org/10.3390/cancers13092241
Thompson EL, Hu JJ, Niedernhofer LJ. The Role of Senescent Cells in Acquired Drug Resistance and Secondary Cancer in BRAFi-Treated Melanoma. Cancers. 2021; 13(9):2241. https://doi.org/10.3390/cancers13092241
Chicago/Turabian StyleThompson, Elizabeth L., Jiayi J. Hu, and Laura J. Niedernhofer. 2021. "The Role of Senescent Cells in Acquired Drug Resistance and Secondary Cancer in BRAFi-Treated Melanoma" Cancers 13, no. 9: 2241. https://doi.org/10.3390/cancers13092241