
Homologous Recombination Repair Deficiency and Implications for Tumor Immunogenicity

 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
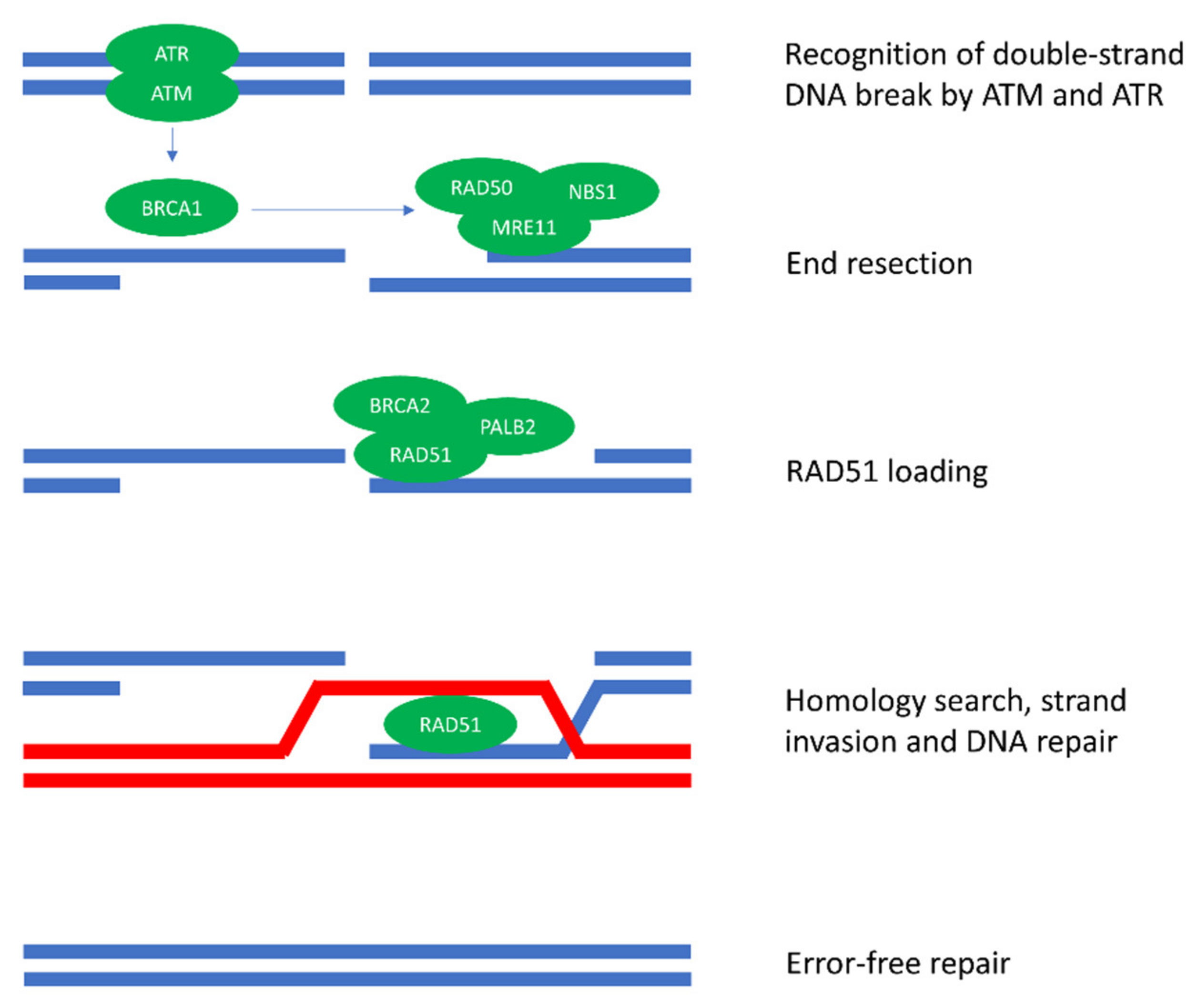
1. Introduction
2. How HRD Influences Antitumor Immunity
2.1. Tumor Mutational Burden and Neoantigen Load
2.2. Copy Number Variations
2.3. STING Pathway
3. The Tumor Immune Microenvironment in BRCA-Inactivated Tumors
3.1. Breast Cancer
3.2. Ovarian Cancer
3.3. Prostate Cancer
- T cells are key players in antitumor immunity. They are able to selectively target cancer cells following recognition of non-self-antigens. T cells, characterized by the expression of CD3, can be subdivided into cytolytic T cells (CD8+), helper T cells (CD4+), and regulatory T cells (CD4+FoxP3+). While cytolytic T cells and helper T cells play an important role in tumor immunosurveillance, regulatory T cells suppress antitumor immunity. Studies in various cancer types indicate that high intratumoral CD8+ T cell density is associated with favorable outcomes to checkpoint inhibitor therapy [62,63]. Nevertheless, the sole presence of CD8+ T cells does not necessarily indicate an active immune response. Immune activity can be inhibited by a lack of antigen presentation or by the presence of immune suppressive cells, cytokines, or inhibitory checkpoint molecules.
- B cells play a major role in antibody-mediated immunity. Although their role in antitumor immunity is not completely understood, recent data suggest that B cells play a role in antitumor immunity and promote checkpoint inhibitor efficacy [64].
- Natural killer cells are innate immune cells with a cytolytic function.
- Checkpoint molecules play an important role in regulating immune responses. PD-L1, PD-1, and LAG-3 are all inhibitory checkpoint molecules. Activation of these checkpoints suppresses immune cell activation. In some cancer types, PD-L1 expression is associated with a response to PD-(L)1 inhibitors [65]. In NSCLC and urothelial cancer, PD-L1 expression is used for treatment stratification.
3.4. Summary
4. Checkpoint Inhibitor Therapy in BRCA-Inactivated Tumors
4.1. Tumor Types with Low Sensitivity to Checkpoint Inhibitor Monotherapy
4.1.1. Breast Cancer
4.1.2. Ovarian Cancer
4.1.3. Prostate Cancer
4.2. Tumor Types Responsive to Checkpoint Inhibitor Monotherapy
4.2.1. Urothelial Cancer
4.2.2. Other Cancer Types
4.3. Pan-Cancer Analyses
4.4. Summary
5. Combining Checkpoint Inhibitors with DNA-Damaging Agents
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.-E.; Malki, M.I. DNA damage/repair management in cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Martens, J.W.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef]
- Pilarski, R. The Role of BRCA Testing in hereditary pancreatic and prostate cancer families. Am. Soc. Clin. Oncol. Educ. B 2019, 39, 79–86. [Google Scholar] [CrossRef]
- Easton, D. Cancer risks in BRCA2 mutation carriers: The breast cancer linkage consortium. J. Natl. Cancer Inst. 1999, 91, 1310–1316. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of homologous recombination–related gene mutations across multiple cancer types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loibl, S.; Weber, K.E.; Timms, K.M.; Elkin, E.P.; Hahnen, E.; Fasching, P.A.; Lederer, B.; Denkert, C.; Schneeweiss, A.; Braun, S.; et al. Survival analysis of carboplatin added to an anthracycline/taxane-based neoadjuvant chemotherapy and HRD score as predictor of response—final results from GeparSixto. Ann. Oncol. 2018, 29, 2341–2347. [Google Scholar] [CrossRef] [PubMed]
- Safra, T.; Rogowski, O.; Muggia, F.M. The effect of germ-line BRCA mutations on response to chemotherapy and outcome of recurrent ovarian cancer. Int. J. Gynecol. Cancer 2014, 24, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Mota, J.M.; Barnett, E.; Nauseef, J.T.; Nguyen, B.; Stopsack, K.H.; Wibmer, A.; Flynn, J.R.; Heller, G.; Danila, D.C.; Rathkopf, D.; et al. Platinum-based chemotherapy in metastatic prostate cancer with DNA repair gene Alterations. JCO Precis. Oncol. 2020, 4, 355–366. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; de Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/ mismatch repair–deficient cancer: Results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Kochupurakkal, B.; Izarzugaza, J.M.G.; Eklund, A.C.; Li, Y.; Liu, J.; Szallasi, Z.; Matulonis, U.A.; Richardson, A.L.; Iglehart, J.D.; et al. Tumor Mutation Burden Forecasts Outcome in Ovarian Cancer with BRCA1 or BRCA2 Mutations. PLoS ONE 2013, 8, e80023. [Google Scholar] [CrossRef] [Green Version]
- Przybytkowski, E.; Davis, T.; Hosny, A.; Eismann, J.; Matulonis, U.A.; Wulf, G.M.; Nabavi, S. An immune-centric exploration of BRCA1 and BRCA2 germline mutation related breast and ovarian cancers. BMC Cancer 2020, 20, 197. [Google Scholar] [CrossRef] [PubMed]
- Van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef] [Green Version]
- Nolan, E.; Savas, P.; Policheni, A.N.; Darcy, P.K.; Vaillant, F.; Mintoff, C.P.; Dushyanthen, S.; Mansour, M.; Pang, J.M.B.; Fox, S.B.; et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci. Transl. Med. 2017, 9, eaal4922. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.X.; Leong, C.O. Association of BRCA1- And BRCA2-deficiency with mutation burden, expression of PD-L1/ PD-1, immune infiltrates, and T cell-inflamed signature in breast cancer. PLoS ONE 2019, 14, e0215381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraya, A.A.; Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; Pluta, J.; Rech, A.J.; Dorfman, L.M.; Lunceford, N.; Barrett, A.; Mitra, N.; et al. Genomic signatures predict the immunogenicity of BRCA-deficient breast cancer. Clin. Cancer Res. 2019, 25, 4363–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wu, L.; Shou, T.; Jiang, B.; Zhuang, L.; Li, K.; Tan, X.; Guo, C.; Guo, W.; Guan, Y.; et al. Abstract 1369: Analysis of association between homologous recombination deficiency and tumor mutational burden in solid tumors. In Proceedings of the Cancer Research, American Association for Cancer Research (AACR), Chicago, IL, USA, 14–18 April 2018; Volume 78, p. 1369. [Google Scholar]
- Chae, Y.K.; Anker, J.F.; Bais, P.; Namburi, S.; Giles, F.J.; Chuang, J.H. Mutations in DNA repair genes are associated with increased neo-antigen load and activated T cell infiltration in lung adenocarcinoma. Oncotarget 2018, 9, 7949–7960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Geukes Foppen, M.H.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Dirk Iglehart, J.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-Mccune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, N.A.; Frock, R.L.; Menghi, F.; Duffey, E.E.; Panday, A.; Camacho, V.; Hasty, E.P.; Liu, E.T.; Alt, F.W.; Scully, R. Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature 2017, 551, 590–595. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Cha, H.; Kim, J.; Park, W.Y.; La Choi, Y.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Lee, S.H. Genomic scoring to determine clinical benefit of immunotherapy by targeted sequencing. Eur. J. Cancer 2019, 120, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Piro, G.; Carbone, C.; Carbognin, L.; Pilotto, S.; Ciccarese, C.; Iacovelli, R.; Milella, M.; Bria, E.; Tortora, G. Revising PTEN in the Era of Immunotherapy: New Perspectives for an Old Story. Cancers 2019, 11, 1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, S.C.; Baylot, V.; Felsher, D.W. MYC: Master Regulator of Immune Privilege. Trends Immunol. 2017, 38, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; Gajewski, T.F. Molecular pathways: Targeting the Stimulator of Interferon Genes (STING) in the immunotherapy of cancer. Clin. Cancer Res. 2015, 21, 4774–4779. [Google Scholar] [CrossRef] [Green Version]
- Pantelidou, C.; Sonzogni, O.; Taveira, M.D.O.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. Parp inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral sting pathway activation in brca-deficient models of triple-negative breast cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.Y.; Tran, J.L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018, 564, 439–443. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.M.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-dependent innate immune signaling by s-phase-specific DNA damage in breast cancer. J. Natl. Cancer Inst. 2017, 109, djw199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Green, A.R.; Aleskandarany, M.A.; Ali, R.; Hodgson, E.G.; Atabani, S.; De Souza, K.; Rakha, E.A.; Ellis, I.O.; Madhusudan, S. Clinical impact of tumor DNA repair expression and T-cell infiltration in breast cancers. Cancer Immunol. Res. 2017, 5, 292–299. [Google Scholar] [CrossRef] [Green Version]
- Solinas, C.; Marcoux, D.; Garaud, S.; Vitória, J.R.; Van den Eynden, G.; de Wind, A.; De Silva, P.; Boisson, A.; Craciun, L.; Larsimont, D.; et al. BRCA gene mutations do not shape the extent and organization of tumor infiltrating lymphocytes in triple negative breast cancer. Cancer Lett. 2019, 450, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Sobral-Leite, M.; Van De Vijver, K.; Michaut, M.; Van Der Linden, R.; Hooijer, G.K.J.; Horlings, H.M.; Severson, T.M.; Mulligan, A.M.; Weerasooriya, N.; Sanders, J.; et al. Assessment of PD-L1 expression across breast cancer molecular subtypes, in relation to mutation rate, BRCA1-like status, tumor-infiltrating immune cells and survival. Oncoimmunology 2018, 7, e1509820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstein, R.M.; Krishna, C.; Ma, X.; Pei, X.; Lee, K.-W.; Makarov, V.; Kuo, F.; Chung, J.; Srivastava, R.M.; Purohit, T.A.; et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat. Cancer 2020, 1, 1188–1203. [Google Scholar] [CrossRef]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B.; Tinker, A.V.; Lee, C.H.; Subramanian, S.; Van De Rijn, M.; Turbin, D.; Kalloger, S.; Han, G.; Ceballos, K.; Cadungog, M.G.; et al. Intraepithelial T cells and prognosis in ovarian carcinoma: Novel associations with stage, tumor type, and BRCA1 loss. Mod. Pathol. 2009, 22, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Wieser, V.; Gaugg, I.; Fleischer, M.; Shivalingaiah, G.; Wenzel, S.; Sprung, S.; Lax, S.F.; Zeimet, A.G.; Fiegl, H.; Marth, C. BRCA1/2 and TP53 mutation status associates with PD-1 and PD-L1 expression in ovarian cancer. Oncotarget 2018, 9, 17501–17511. [Google Scholar] [CrossRef] [Green Version]
- McAlpine, J.N.; Porter, H.; Köbel, M.; Nelson, B.H.; Prentice, L.M.; Kalloger, S.E.; Senz, J.; Milne, K.; Ding, J.; Shah, S.P.; et al. BRCA1 and BRCA2 mutations correlate with TP53 abnormalities and presence of immune cell infiltrates in ovarian high-grade serous carcinoma. Mod. Pathol. 2012, 25, 740–750. [Google Scholar] [CrossRef] [Green Version]
- Whitehair, R.; Peres, L.C.; Mills, A.M. Expression of the Immune Checkpoints LAG-3 and PD-L1 in High-grade Serous Ovarian Carcinoma. Int. J. Gynecol. Pathol. 2019, 39, 558–566. [Google Scholar] [CrossRef]
- Dai, Y.; Sun, C.; Feng, Y.; Jia, Q.; Zhu, B. Potent immunogenicity in BRCA1-mutated patients with high-grade serous ovarian carcinoma. J. Cell. Mol. Med. 2018, 22, 3979–3986. [Google Scholar] [CrossRef]
- Jenzer, M.; Keß, P.; Nientiedt, C.; Endris, V.; Kippenberger, M.; Leichsenring, J.; Stögbauer, F.; Haimes, J.; Mishkin, S.; Kudlow, B.; et al. The BRCA2 mutation status shapes the immune phenotype of prostate cancer. Cancer Immunol. Immunother. 2019, 68, 1621–1633. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Aguiar, P.N.; De Mello, R.A.; Hall, P.; Tadokoro, H.; De Lima Lopes, G. PD-L1 expression as a predictive biomarker in advanced non-small-cell lung cancer: Updated survival data. Immunotherapy 2017, 9, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous recombination deficiency (hrd) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Wu, J.; Zhang, Z.; Tang, Y.; Li, X.; Liu, S.; Cao, S.; Li, X. Association between BRCA status and triple-negative breast cancer: A meta-analysis. Front. Pharmacol. 2018, 9, 909. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, A.; Nanda, R. Immune Checkpoint Blockade for Breast Cancer. In Cancer Treatment and Research; Springer International Publishing: New York, NY, USA, 2018; Volume 173, pp. 155–165. [Google Scholar]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Molinero, L.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Diéras, V.; Iwata, H.; Barrios, C.H.; Nechaeva, M.; Nguyen Duc, A.; et al. Atezolizumab and nab-Paclitaxel in Advanced Triple-Negative Breast Cancer: Biomarker Evaluation of the IMpassion130 Study (CHB); Arkhangelsk Regional Clinical. J. Nat. Cancer Inst. 2021, djab004. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.F.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Results from the phase II KEYNOTE-100 study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef]
- Disis, M.L.; Taylor, M.H.; Kelly, K.; Beck, J.T.; Gordon, M.; Moore, K.M.; Patel, M.R.; Chaves, J.; Park, H.; Mita, A.C.; et al. Efficacy and Safety of Avelumab for Patients with Recurrent or Refractory Ovarian Cancer: Phase 1 b Results from the JAVELIN Solid Tumor Trial. JAMA Oncol. 2019, 5, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Spragg, S.E.; Ciccone, M.A.; Blake, E.A.; Ricker, C.; Pham, H.Q.; Roman, L.D. Nivolumab use for BRCA gene mutation carriers with recurrent epithelial ovarian cancer: A case series. Gynecol. Oncol. Rep. 2018, 25, 98–101. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: Multicohort, open-label phase II KEYNOTE-199 study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Flechon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M.; et al. Initial results from a phase II study of nivolumab (NIVO) plus ipilimumab (IPI) for the treatment of metastatic castration-resistant prostate cancer (mCRPC.; CheckMate 650). J. Clin. Oncol. 2019, 37, 142. [Google Scholar] [CrossRef]
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Fléchon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M.; et al. Nivolumab Plus Ipilimumab for Metastatic Castration-Resistant Prostate Cancer: Preliminary Analysis of Patients in the CheckMate 650 Trial. Cancer Cell 2020, 38, 489–499.e3. [Google Scholar] [CrossRef]
- Fradet, Y.; Bellmunt, J.; Vaughn, D.J.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; Necchi, A.; et al. Randomized phase III KEYNOTE-045 trial of pembrolizumab versus paclitaxel, docetaxel, or vinflunine in recurrent advanced urothelial cancer: Results of >2 years of follow-up. Ann. Oncol. 2019, 30, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Alva, A.; Csőszi, T.; Ozguroglu, M.; Matsubara, N.; Geczi, L.; Cheng, S.Y.; Fradet, Y.; Oudard, S.; Vulsteke, C.; Morales Barrera, R.; et al. LBA23—Pembrolizumab (p) combined with chemotherapy (C) vs C alone as first-line (1 L) therapy for advanced urothelial carcinoma (UC): KEYNOTE-361. Ann. Oncol. 2020, 31 (Suppl. S4), S1142–S1215. [Google Scholar] [CrossRef]
- Powles, T.; van der Heijden, M.S.; Castellano, D.; Galsky, M.D.; Loriot, Y.; Petrylak, D.P.; Ogawa, O.; Park, S.H.; Lee, J.L.; De Giorgi, U.; et al. Durvalumab alone and durvalumab plus tremelimumab versus chemotherapy in previously untreated patients with unresectable, locally advanced or metastatic urothelial carcinoma (DANUBE): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1574–1588. [Google Scholar] [CrossRef]
- Teo, M.Y.; Seier, K.; Ostrovnaya, I.; Regazzi, A.M.; Kania, B.E.; Moran, M.M.; Cipolla, C.K.; Bluth, M.J.; Chaim, J.; Al-Ahmadie, H.; et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J. Clin. Oncol. 2018, 36, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Hsiehchen, D.; Hsieh, A.; Samstein, R.M.; Wang, T.; Morris, L.G.T.; Correspondence, H.Z.; Lu, T.; Beg, M.S.; Gerber, D.E.; Zhu, H. DNA Repair Gene Mutations as Predictors of Immune Checkpoint Inhibitor Response beyond Tumor Mutation Burden ll. Cell Rep Med. 2020, 1, 100034. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Gordon, M.; Veneris, J.; Braiteh, F.; Balmanoukian, A.; Eder, J.P.; Oaknin, A.; Hamilton, E.; Wang, Y.; Sarkar, I.; et al. Safety, clinical activity and biomarker assessments of atezolizumab from a Phase I study in advanced/recurrent ovarian and uterine cancers. Gynecol. Oncol. 2019, 154, 314–322. [Google Scholar] [CrossRef]
- Boudadi, K.; Suzman, D.L.; Anagnostou, V.; Fu, W.; Luber, B.; Wang, H.; Niknafs, N.; White, J.R.; Silberstein, J.L.; Sullivan, R.; et al. Ipilimumab plus nivolumab and DNA-repair defects in AR-V7-expressing metastatic prostate cancer. Oncotarget 2018, 9, 28561–28571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, P.; Yang, D.; Wang, H.; Cui, X.; Si, X.; Zhang, X.; Zhang, L. Relationship between the efficacy of immunotherapy and characteristics of specific tumor mutation genes in non-small cell lung cancer patients. Thorac. Cancer 2020, 11, 1647–1654. [Google Scholar] [CrossRef] [PubMed]
- Labriola, M.K.; Zhu, J.; Gupta, R.; McCall, S.; Jackson, J.; Kong, E.F.; White, J.R.; Cerqueira, G.; Gerding, K.; Simmons, J.K.; et al. Characterization of tumor mutation burden, PD-L1 and DNA repair genes to assess relationship to immune checkpoint inhibitors response in metastatic renal cell carcinoma. J. Immunother. Cancer 2020, 8, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, F.; Zhang, Y.; Chen, S.; Weng, X.; Rao, Y.; Fang, H. Mechanism and current progress of Poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed. Pharmacother. 2020, 123, 109661. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPI triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCANEss. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Robillard, L.; Nguyen, M.; Loehr, A.; Orsulic, S.; Kristeleit, R.S.; Lin, K.; Raponi, M.; Harding, T.C.; Simmons, A.D. Abstract 3650: Preclinical evaluation of the PARP inhibitor rucaparib in combination with PD-1 and PD-L1 inhibition in a syngeneic BRCA1 mutant ovarian cancer model. In Proceedings of the Immunology, American Association for Cancer Research, Washington, DC, USA, 1–5 April 2017; Volume 77, p. 3650. [Google Scholar]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [Green Version]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Reference | Tumor Type | N | Genes | Treatment | Results 1 | |
|---|---|---|---|---|---|---|
| Total | Mut | |||||
| [70] | TNBC | 612 | 89 | Pathogenic germline or somatic BRCA1/2 variants, zygosity status not assessed | Atezolizumab + nab-paclitaxel | PFS: hazard ratio 1.07, 95% CI 0.77–1.49 OS: hazard ratio 1.07, 95% CI 0.71–1.62 |
| [71] | TNBC | 49 | 25 | BRCA1-like genomic copy number profiles | Nivolumab with or without induction chemotherapy or irradiation | Lower ORR in BRCA1-like patients (p < 0.05) |
| [72] | Ovarian cancer | 46 | 8 | Pathogenic germline BRCA1/2 variants, zygosity status not assessed | Avelumab | ORR: 12.5% (1/8) in BRCA-mut vs 7.9% (3/38) in BRCA-WT |
| [73] | Ovarian or fallopian tubal cancer | 6 | 6 | Pathogenic germline BRCA1/2 variants, zygosity status not assessed | Nivolumab | ORR: 76% (3/6 CR, 1/6 PR, 2/6 PD) |
| [74] | Ovarian or uterine cancer | 25 | 2 | Pathogenic germline BRCA1 variants | Atezolizumab | ORR: Both had PD |
| [75] | CRPC | 153 | 19 | Pathogenic homozygous BRCA1/2 or ATM aberrations | Pembrolizumab | ORR: 11% (2/19) in patients with BRCA1/2 or ATM aberrations and 3% (4/124) in patients without HR aberrations |
| [76] | CRPC | 28 | 5 | Pathogenic homozygous aberrations in BRCA2, ATM, CDK12, or FANCA | Ipilimumab + nivolumab | ORR: 50% (3/6) in DDR-mut vs 22.6% (7/31) in DDR-WT. Of note, responding patients in the DDR group had mutations in BRCA2 or FANCA |
| [77] | CRPC with AR-V7 expression | 15 | 6 | Pathogenic mutations in BRCA2 (3), ATM (2), ERCC4 (1)2, LOH in two BRCA2-mut patients | Ipilimumab + nivolumab | ORR: 40% (2/5) in DDR-mut vs 0% (0/3) in DDR-WT (p = 0.46) PSA response: 33% (2/6) vs 0% (0/9) (p = 014) PFS: hazard ratio = 0.31, 95% CI 0.10–0.92, p = 0.01 OS: hazard ratio = 0.41, 95% CI 0.14–1.21, p = 0.1 |
| [78] | Urothelial cancer | 60 | 15 | Pathogenic alterations in BRCA1/2 (3) and other DDR genes (12; ATM, POLE, ERCC2, CHEK2, FANCA, and MSH2, MSH6). Zygosity status n/a | Anti-PD-(L)1 | ORR: 80% (12/15) and 19% (6/32) in patients with deleterious DDR alterations and no DDR alterations, resp. PFS: Median PFS NR3 and 2.9 months, resp |
| [79] | NSCLC | 44 | 9 | BRCA1/2 mutations. Zygosity status and pathogenicity n/a | Anti-PD-(L)1 | 10% and 29% of patients with and without durable benefit resp, harbored a mutation in BRCA1/2 |
| [80] | Renal cell carcinoma | 34 | 12 | BRCA1/2 mutations. Zygosity status and pathogenicity n/a | Anti-PD-1 alone (32) or combined with anti-CTLA-4 (2) | 38% (6/16) of patients with disease control vs. 33% (6/18) of patients with PD had a mutation in an BRCA1/2 |
| [81] | Metastatic melanoma | 38 | 7 | BRCA2 mutations. Zygosity status and pathogenicity n/a | Anti-PD-1 | BRCA2 mutation in 28% (6/21) of responders vs. 6% (1/17) of non-responders |
| [82] | Various solid tumors | 1661 | 335 | ARID1 A, BLM, BRCA2, MRE11, NBN, RAD50, RAD51/B/D, RAD52, RAD54 L, XRCC2 Zygosity status and pathogenicity n/a | Anti-CTLA-4 (9%), anti-PD-(L)1 (76%), or both (16%) | OS: Median OS 41 months in HR-mut vs 16 months in HR-WT Adj hazard ratio4 = 1.39, 95% CI 1.15–1.70, p = 0.022 |
| [54] | Various tumors | 2185 | 95 | Pathogenic somatic or germline BRCA1 (28) or BRCA2 (67) mutations. Zygosity statis n/a | Anti-PD-(L)1, CTLA-4 or a combination | OS BRCA1: Hazard ratio 0.76, 95% CI 0.48–1.54, p = 0.45 OS BRCA2: Hazard ratio 0.48, 95% CI 0.29–0.80 Adj hazard ratio5 = 0.50, 95% CI 0.30–0.83, p = 0.008 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Wilpe, S.; Tolmeijer, S.H.; Koornstra, R.H.T.; de Vries, I.J.M.; Gerritsen, W.R.; Ligtenberg, M.; Mehra, N. Homologous Recombination Repair Deficiency and Implications for Tumor Immunogenicity. Cancers 2021, 13, 2249. https://doi.org/10.3390/cancers13092249
van Wilpe S, Tolmeijer SH, Koornstra RHT, de Vries IJM, Gerritsen WR, Ligtenberg M, Mehra N. Homologous Recombination Repair Deficiency and Implications for Tumor Immunogenicity. Cancers. 2021; 13(9):2249. https://doi.org/10.3390/cancers13092249
Chicago/Turabian Stylevan Wilpe, Sandra, Sofie H. Tolmeijer, Rutger H. T. Koornstra, I. Jolanda M. de Vries, Winald R. Gerritsen, Marjolijn Ligtenberg, and Niven Mehra. 2021. "Homologous Recombination Repair Deficiency and Implications for Tumor Immunogenicity" Cancers 13, no. 9: 2249. https://doi.org/10.3390/cancers13092249
APA Stylevan Wilpe, S., Tolmeijer, S. H., Koornstra, R. H. T., de Vries, I. J. M., Gerritsen, W. R., Ligtenberg, M., & Mehra, N. (2021). Homologous Recombination Repair Deficiency and Implications for Tumor Immunogenicity. Cancers, 13(9), 2249. https://doi.org/10.3390/cancers13092249