
IGF2 Mediates Resistance to Isoform-Selective-Inhibitors of the PI3K in HPV Positive Head and Neck Cancer

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Cell Lines
2.2. IC50 and Synergy Assay
2.3. Cell Proliferation Assay
2.4. Cell Cycle
2.5. RNAseq
2.6. Real-Time PCR
2.7. siRNA Transfection
2.8. Western Blotting
2.9. Immunohistochemistry (IHC) and Immunofluorescence (IF)
2.10. Tumor Ex Vivo Analysis (TEVA)
2.11. Establishment of Tumor Xenografts and Studies in Mice
2.12. Antibodies
2.13. Drugs and Reagents
2.14. Statistics
2.15. Study Approval
3. Results
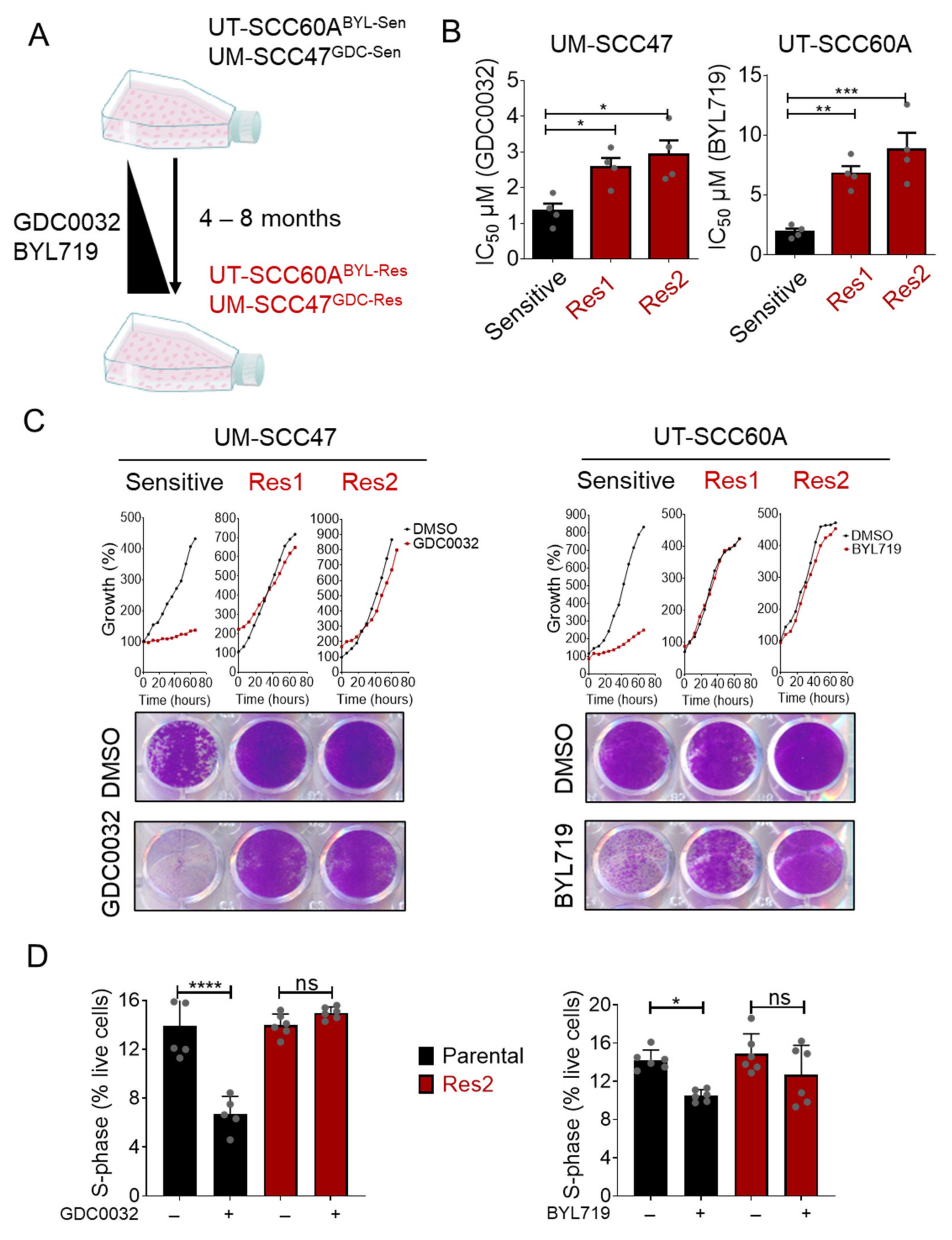
3.1. Generation of BYL719- and GDC0032-Acquired Resistance HNCHPV+ Cell Lines
3.2. FOXO Signaling Pathway Is Activated in isiPI3K-Resistant Cells
3.3. Overexpression of IGF2 Limits the Vulnerability to isiPI3K
3.4. IGF1R Inhibition Synergizes with isiPI3K to Inhibit Tumor Cells
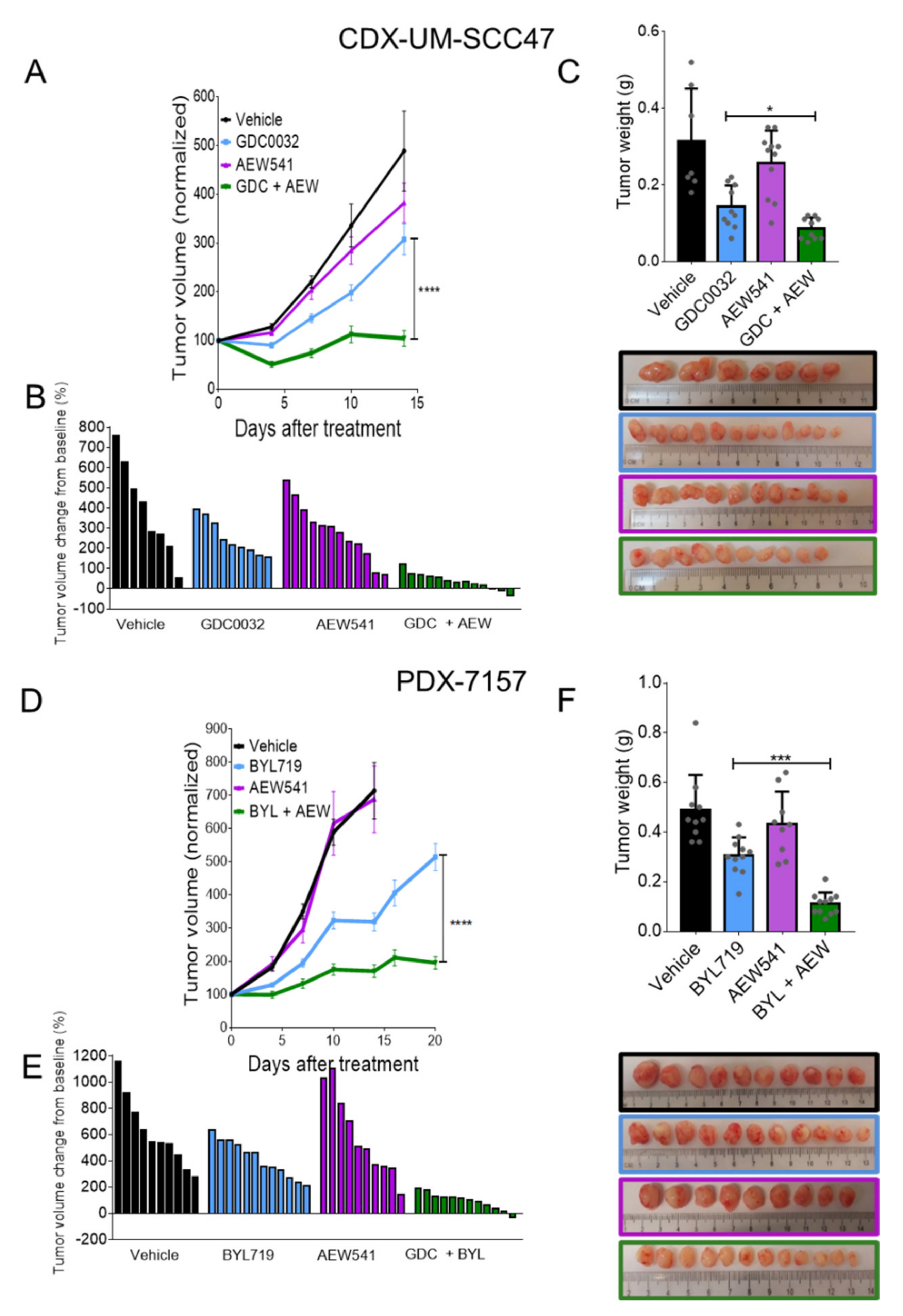
3.5. Dual Treatment with isiPI3K and AEW541 Induced Growth Arrest in PDXHPV+ Ex Vivo
3.6. Dual Treatment with isiPI3K and AEW541 Induced Tumor Growth Arrest in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cadoni, G.; Giraldi, L.; Petrelli, L.; Pandolfini, M.; Giuliani, M.; Paludetti, G.; Pastorino, R.; Leoncini, E.; Arzani, D.; Almadori, G.; et al. Prognostic factors in head and neck cancer: A 10-year retrospective analysis in a single-institution in Italy. Acta Otorhinolaryngol. Ital. 2017, 37, 458–466. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Kawakita, D.; Matsuo, K. Alcohol and head and neck cancer. Cancer Metastasis Rev. 2017, 36, 425–434. [Google Scholar] [CrossRef]
- Chung, C.H.; Bagheri, A.; D’Souza, G. Epidemiology of oral human papillomavirus infection. Oral Oncol. 2014, 50, 364–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Kiess, A.; Chung, C.H. Emerging biomarkers in head and neck cancer in the era of genomics. Nat. Rev. Clin. Oncol. 2015, 12, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human Papillomavirus and Rising Oropharyngeal Cancer Incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef]
- Dayyani, F.; Etzel, C.J.; Liu, M.; Ho, C.-H.; Lippman, S.M.; Tsao, A.S. Meta-analysis of the impact of human papillomavirus (HPV) on cancer risk and overall survival in head and neck squamous cell carcinomas (HNSCC). Head Neck Oncol. 2010, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Lassen, P.; Eriksen, J.G.; Hamilton-Dutoit, S.; Tramm, T.; Alsner, J.; Overgaard, J. Effect of HPV-Associated p16INK4A Expression on Response to Radiotherapy and Survival in Squamous Cell Carcinoma of the Head and Neck. J. Clin. Oncol. 2009, 27, 1992–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausen, H.Z. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Paz, I.B.; Cook, N.; Odom-Maryon, T.; Xie, Y.; Wilczynski, S.P. Human papillomavirus (HPV) in head and neck cancer. An association of HPV 16 with squamous cell carcinoma of Waldeyer’s tonsillar ring. Cancer 1997, 79, 595–604. [Google Scholar] [CrossRef]
- Bodily, J.; Laimins, L.A. Persistence of human papillomavirus infection: Keys to malignant progression. Trends Microbiol. 2011, 19, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Syrjanen, S. Human papillomaviruses in head and neck carcinomas. N. Engl. J. Med. 2007, 356, 1993–1995. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of Human Papillomavirus–Positive Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [Green Version]
- Pinatti, L.; Walline, H.; Carey, T. Human Papillomavirus Genome Integration and Head and Neck Cancer. J. Dent. Res. 2017, 97, 691–700. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar]
- Sano, T.; Oyama, T.; Kashiwabara, K.; Fukuda, T.; Nakajima, T. Expression Status of p16 Protein Is Associated with Human Papillomavirus Oncogenic Potential in Cervical and Genital Lesions. Am. J. Pathol. 1998, 153, 1741–1748. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nat. Cell Biol. 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL Mediates Resistance to PI3Kα Inhibition by Activating the EGFR/PKC/mTOR Axis in Head and Neck and Esophageal Squamous Cell Carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [Green Version]
- Elkabets, M.; Vora, S.; Juric, D.; Morse, N.; Mino-Kenudson, M.; Muranen, T.; Tao, J.; Campos, A.B.; Rodon, J.; Ibrahim, Y.H.; et al. mTORC1 Inhibition Is Required for Sensitivity to PI3K p110α Inhibitors inPIK3CA-Mutant Breast Cancer. Sci. Transl. Med. 2013, 5, 196ra99. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the Novel and Specific PI3Kα Inhibitor NVP-BYL719 and Development of the Patient Stratification Strategy for Clinical Trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef] [Green Version]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorgan. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef] [PubMed]
- Zumsteg, Z.S.; Morse, N.; Krigsfeld, G.S.; Gupta, G.; Higginson, D.S.; Lee, N.Y.; Morris, L.G.T.; Ganly, I.; Shiao, S.L.; Powell, S.N.; et al. Taselisib (GDC-0032), a Potent β-Sparing Small Molecule Inhibitor of PI3K, Radiosensitizes Head and Neck Squamous Carcinomas Containing Activating PIK3CA Alterations. Clin. Cancer Res. 2016, 22, 2009–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, G.J.; Kacew, A.; Chau, N.G.; Shivdasani, P.; Lorch, J.H.; Uppaluri, R.; Haddad, R.I.; MacConaill, L.E. Improved outcomes in PI3K-pathway-altered metastatic HPV oropharyngeal cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimeno, A.; Bauman, J.E.; Weissman, C.; Adkins, D.; Schnadig, I.; Beauregard, P.; Bowles, D.W.; Spira, A.; Levy, B.; Seetharamu, N.; et al. A randomized, phase 2 trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Oral Oncol. 2015, 51, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, Y.; Inada-Inoue, M.; Mitsuma, A.; Yoshino, T.; Ohtsu, A.; Suenaga, N.; Sato, M.; Kakizume, T.; Robson, M.; Quadt, C.; et al. Phase I dose-escalation study of buparlisib ( BKM 120), an oral pan-class I PI 3K inhibitor, in Japanese patients with advanced solid tumors. Cancer Sci. 2014, 105, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roper, J.; Richardson, M.P.; Wang, W.V.; Richard, L.G.; Chen, W.; Coffee, E.M.; Sinnamon, M.J.; Lee, L.; Chen, P.-C.; Bronson, R.T.; et al. The Dual PI3K/mTOR Inhibitor NVP-BEZ235 Induces Tumor Regression in a Genetically Engineered Mouse Model of PIK3CA Wild-Type Colorectal Cancer. PLoS ONE 2011, 6, e25132. [Google Scholar] [CrossRef] [Green Version]
- Mayer, I.A.; Abramson, V.G.; Formisano, L.; Balko, J.M.; Estrada, M.V.; Sanders, M.E.; Juric, D.; Solit, D.; Berger, M.F.; Won, H.H.; et al. A Phase Ib Study of Alpelisib (BYL719), a PI3Kα-Specific Inhibitor, with Letrozole in ER+/HER2− Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 26–34. [Google Scholar] [CrossRef] [Green Version]
- US Food and Drug Administration. FDA approves alpelisib for metastatic breast cancer. Case Med. Res. 2019, 2–3. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-Kinase α–Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Iwasa, S.; Takahashi, S.; Saka, H.; Kakizume, T.; Natsume, K.; Suenaga, N.; Quadt, C.; Yamada, Y. Phase I study of alpelisib (BYL719), an α-specific PI3K inhibitor, in Japanese patients with advanced solid tumors. Cancer Sci. 2019, 110, 1021–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric, D.; Krop, I.; Ramanathan, R.K.; Wilson, T.R.; Ware, J.A.; Bohorquez, S.M.S.; Savage, H.M.; Sampath, D.; Salphati, L.; Lin, R.S.; et al. Phase I Dose-Escalation Study of Taselisib, an Oral PI3K Inhibitor, in Patients with Advanced Solid Tumors. Cancer Discov. 2017, 7, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhaveri, K.; Chang, M.T.; Juric, D.; Saura, C.; Gambardella, V.; Melnyk, A.; Patel, M.R.; Ribrag, V.; Ma, C.X.; Aljumaily, R.; et al. Phase I Basket Study of Taselisib, an Isoform-Selective PI3K Inhibitor, in Patients with PIK3CA-Mutant Cancers. Clin. Cancer Res. 2021, 27, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Yegodayev, K.M.; Novoplansky, O.; Golden, A.; Prasad, M.; Levin, L.; Jagadeeshan, S.; Zorea, J.; Dimitstein, O.; Joshua, B.-Z.; Cohen, L.; et al. TGF-Beta-Activated Cancer-Associated Fibroblasts Limit Cetuximab Efficacy in Preclinical Models of Head and Neck Cancer. Cancers 2020, 12, 339. [Google Scholar] [CrossRef] [Green Version]
- Sklarz, M.; Levin, L.; Gordon, M.; Chalifa-Caspi, V. NeatSeq-Flow: A Lightweight High-Throughput Sequencing Workflow Platform for Non-Programmers and Programmers Alike. bioRxiv 2017. Available online: https://www.biorxiv.org/content/10.1101/173005v3 (accessed on 24 March 2021). [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Parrish, N.; Hormozdiari, F.; Eskin, E. Assembly of non-unique insertion content using next-generation sequencing. In Bioinformatics: The Impact of Accurate Quantification on Proteomic and Genetic Analysis and Research, 1st ed.; Liu, Y., Ed.; Apple Academic Press: New York, NY, USA, 2014; pp. 21–40. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Jouhi, L.; Datta, N.; Renkonen, S.; Atula, T.; Mäkitie, A.; Haglund, C.; Ahmed, A.; Syrjänen, S.; Grénman, R.; Auvinen, E.; et al. Expression of toll-like receptors in HPV-positive and HPV-negative oropharyngeal squamous cell carcinoma—an in vivo and in vitro study. Tumor Biol. 2015, 36, 7755–7764. [Google Scholar] [CrossRef]
- Lin, A.; Piao, H.-L.; Zhuang, L.; Sarbassov, S.D.; Ma, L.; Gan, B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacologic inhibition of the PI3K-AKT pathway. Cancer Res. 2014, 74, 1682–1693. [Google Scholar] [CrossRef] [Green Version]
- Tenbaum, S.P.; Ordóñez-Morán, P.; Puig, I.; Chicote, I.; Arqués, O.; Landolfi, S.; Fernández, Y.; Herance, J.R.; Gispert, J.D.; Mendizabal, L.; et al. β-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat. Med. 2012, 18, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Brouwer-Visser, J.; Huang, G.S. IGF2 signaling and regulation in cancer. Cytokine Growth Factor Rev. 2015, 26, 371–377. [Google Scholar] [CrossRef]
- Lei, J.; Wang, S.; Kang, W.; Chu, Q.; Liu, Z.; Sun, L.; Ji, Y.; Esteban, C.R.; Yao, Y.; Belmonte, J.C.I.; et al. FOXO3-engineered human mesenchymal progenitor cells efficiently promote cardiac repair after myocardial infarction. Protein Cell 2021, 12, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for Drug Synergy in Complex Dose–Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvino, C.L.; Ong, S.C.; McNeil, K.A.; Delaine, C.; Booker, G.W.; Wallace, J.C.; Forbes, B.E. Understanding the Mechanism of Insulin and Insulin-Like Growth Factor (IGF) Receptor Activation by IGF-II. PLoS ONE 2011, 6, e27488. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Livingstone, C. IGF2 and cancer. Endocr. Relat. Cancer 2013, 20, R321–R339. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Prasad, M.; Kundu, K.; Cohen, L.; Yegodayev, K.M.; Zorea, J.; Joshua, B.-Z.; Lasry, B.; Dimitstein, O.; Bahat-Dinur, A.; et al. Tumor Tissue Explant Culture of Patient-Derived Xenograft as Potential Prioritization Tool for Targeted Therapy. Front. Oncol. 2019, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paddock, M.N.; Field, S.J.; Cantley, L.C. Treating cancer with phosphatidylinositol-3-kinase inhibitors: Increasing efficacy and overcoming resistance. J. Lipid Res. 2019, 60, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Bauer, T.M.; Patel, M.R.; Infante, J.R. Targeting PI3 kinase in cancer. Pharmacol. Ther. 2015, 146, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Elmenier, F.M.; Lasheen, D.S.; Abouzid, K.A. Phosphatidylinositol 3 kinase (PI3K) inhibitors as new weapon to combat cancer. Eur. J. Med. Chem. 2019, 183, 111718. [Google Scholar] [CrossRef] [PubMed]
- Ilagan, E.; Manning, B.D. Emerging Role of mTOR in the Response to Cancer Therapeutics. Trends Cancer 2016, 2, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bago, R.; Sommer, E.; Castel, P.; Crafter, C.; Bailey, F.P.; Shpiro, N.; Baselga, J.; Cross, D.; Eyers, P.A.; Alessi, D.R. The hVps34- SGK 3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC 1 and tumour growth. EMBO J. 2016, 35, 1902–1922. [Google Scholar] [CrossRef] [PubMed]
- Castel, P.; Ellis, H.; Bago, R.; Toska, E.; Razavi, P.; Carmona, F.J.; Kannan, S.; Verma, C.S.; Dickler, M.; Chandarlapaty, S.; et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kα Inhibition. Cancer Cell 2016, 30, 229–242. [Google Scholar] [CrossRef] [Green Version]
- Ilic, N.; Utermark, T.; Widlund, H.R.; Roberts, T.M. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc. Natl. Acad. Sci. USA 2011, 108, E699–E708. [Google Scholar] [CrossRef] [Green Version]
- Michmerhuizen, N.L.; Leonard, E.; Kulkarni, A.; Brenner, J.C. Differential compensation mechanisms define resistance to PI3K inhibitors in PIK3CA amplified HNSCC. Otorhinolaryngol. Neck Surg. 2016, 1, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Badarni, M.; Prasad, M.; Balaban, N.; Zorea, J.; Yegodayev, K.M.; Ben-Zion, J.; Dinur, A.B.; Grénman, R.; Rotblat, B.; Cohen, L.; et al. Repression of AXL expression by AP-1/JNK blockage overcomes resistance to PI3Ka therapy. JCI Insight 2019, 4, e125341. [Google Scholar] [CrossRef] [PubMed]
- Ruicci, K.M.; Meens, J.; Plantinga, P.; Stecho, W.; Pinto, N.; Yoo, J.; Fung, K.; MacNeil, D.; Mymryk, J.S.; Barrett, J.W.; et al. TAM family receptors in conjunction with MAPK signalling are involved in acquired resistance to PI3Kα inhibition in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 217. [Google Scholar] [CrossRef]
- Meister, K.S.; Godse, N.R.; Khan, N.I.; Hedberg, M.L.; Kemp, C.; Kulkarni, S.; Alvarado, D.; Lavallee, T.; Kim, S.; Grandis, J.R.; et al. HER3 targeting potentiates growth suppressive effects of the PI3K inhibitor BYL719 in pre-clinical models of head and neck squamous cell carcinoma. Sci. Rep. 2019, 9, 9130. [Google Scholar] [CrossRef] [Green Version]
- Han, M.W.; Ryu, I.S.; Lee, J.C.; Kim, S.H.; Chang, H.W.; Lee, Y.S.; Lee, S.; Kim, S.W.; Kim, S.Y. Phosphorylation of PI3K regulatory subunit p85 contributes to resistance against PI3K inhibitors in radioresistant head and neck cancer. Oral Oncol. 2018, 78, 56–63. [Google Scholar] [CrossRef]
- Remer, E.; Badarni, M.; Hikri, E.; Dayan, A.; Levi, L.; Popovtzer, A.; Iraqi, M.; Porgador, A.; Joshua, B.-Z.; Bachar, G.; et al. CDK 4/6 Inhibition Overcomes Acquired and Inherent Resistance to PI3Kα Inhibition in Pre-Clinical Models of Head and Neck Squamous Cell Carcinoma. J. Clin. Med. 2020, 9, 3214. [Google Scholar] [CrossRef]
- Brand, T.M.; Hartmann, S.; Bhola, N.E.; Li, H.; Zeng, Y.; O’Keefe, R.A.; Ranall, M.V.; Bandyopadhyay, S.; Soucheray, M.; Krogan, N.J.; et al. Cross-talk Signaling between HER3 and HPV16 E6 and E7 Mediates Resistance to PI3K Inhibitors in Head and Neck Cancer. Cancer Res. 2018, 78, 2383–2395. [Google Scholar] [CrossRef] [Green Version]
- Pereira, S.S.; Monteiro, M.P.; Costa, M.M.; Moreira, Â.; Alves, M.G.; Oliveira, P.F.; Jarak, I.; Pignatelli, D. IGF2 role in adrenocortical carcinoma biology. Endocrine 2019, 66, 326–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Nogueras, J.; Weiss, E.; Cruz-Correa, M.; Berho, M.; Sands, D.; Wexner, S.; Giardiello, F.M. Positive Correlation of Insulin-Like Growth Factor-II with Proliferating Cell Index in Patients with Colorectal Neoplasia. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1819–1822. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.K.; Huang, Z.; Wen, Y.; Spillman, M.A.; Whitaker, R.S.; Simel, L.R.; Nichols, T.D.; Marks, J.R.; Berchuck, A. Frequent IGF2/H19 Domain Epigenetic Alterations and Elevated IGF2 Expression in Epithelial Ovarian Cancer. Mol. Cancer Res. 2006, 4, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; DeCoteau, J.F.; Geyer, C.; Gao, M.; Cui, H.; Casson, A.G. Loss of imprinting of the insulin-like growth factor II (IGF2) gene in esophageal normal and adenocarcinoma tissues. Carcinogenesis 2009, 30, 2117–2122. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-K.; Weksberg, R.; Minden, M.D.; Squire, J.A. Loss of Imprinting of Human Insulin-like Growth Factor II Gene, IGF2, in Acute Myeloid Leukemia. Biochem. Biophys. Res. Commun. 1997, 231, 466–472. [Google Scholar] [CrossRef]
- Huang, G.S.; Brouwer-Visser, J.; Ramirez, M.J.; Kim, C.H.; Hebert, T.M.; Lin, J.; Arias-Pulido, H.; Qualls, C.R.; Prossnitz, E.R.; Goldberg, G.L.; et al. Insulin-like Growth Factor 2 Expression Modulates Taxol Resistance and Is a Candidate Biomarker for Reduced Disease-Free Survival in Ovarian Cancer. Clin. Cancer Res. 2010, 16, 2999–3010. [Google Scholar] [CrossRef] [Green Version]
- Brouwer-Visser, J.; Lee, J.; Mccullagh, K.; Cossio, M.J.; Wang, Y.; Huang, G.S. Insulin-Like Growth Factor 2 Silencing Restores Taxol Sensitivity in Drug Resistant Ovarian Cancer. PLoS ONE 2014, 9, e100165. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tsao, S.W.; Chan, K.W.; Ludwig, D.L.; Novosyadlyy, R.; Li, Y.Y.; He, Q.Y.; Cheung, A.L. Id1-Induced IGF-II and Its Autocrine/Endocrine Promotion of Esophageal Cancer Progression and Chemoresistance—Implications for IGF-II and IGF-IR–Targeted Therapy. Clin. Cancer Res. 2014, 20, 2651–2662. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, T.; Ogawa, K.; Shiga, K.; Furukawa, T.; Nagase, H.; Hashimoto, S.; Kobayashi, T.; Horii, A. Upregulation of IGF2 is associated with an acquired resistance for cis-diamminedichloroplatinum in human head and neck squamous cell carcinoma. Eur. Arch. Oto-Rhino-Laryngol. 2010, 267, 1599–1606. [Google Scholar] [CrossRef]
- Manabe, T.; Yasuda, H.; Terai, H.; Kagiwada, H.; Hamamoto, J.; Ebisudani, T.; Kobayashi, K.; Masuzawa, K.; Ikemura, S.; Kawada, I.; et al. IGF2 Autocrine-Mediated IGF1R Activation Is a Clinically Relevant Mechanism of Osimertinib Resistance in Lung Cancer. Mol. Cancer Res. 2020, 18, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorea, J.; Prasad, M.; Cohen, L.; Li, N.; Schefzik, R.; Ghosh, S.; Rotblat, B.; Brors, B.; Elkabets, M. IGF1R upregulation confers resistance to isoform-specific inhibitors of PI3K in PIK3CA-driven ovarian cancer. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, E.M.; Kuba, M.G.; Miller, T.W.; Davies, B.R.; Arteaga, C.L. Autocrine IGF-I/insulin receptor axis compensates for inhibition of AKT in ER-positive breast cancer cells with resistance to estrogen deprivation. Breast Cancer Res. 2013, 15, R55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, V.L.; Barnes, D.J.; Sandovici, I.; Constancia, M.; Graham, C.F.; Pezzella, F.; Bühnemann, C.; Carter, E.J.; Hassan, A.B. Igf2 pathway dependency of the Trp53 developmental and tumour phenotypes. EMBO Mol. Med. 2012, 4, 705–718. [Google Scholar] [CrossRef]
- Zhang, L.; Kashanchi, F.; Zhan, Q.; Zhan, S.; Brady, J.N.; Fornace, A.J.; Seth, P.; Helman, L.J. Regulation of insulin-like growth factor II P3 promotor by p53: A potential mechanism for tumorigenesis. Cancer Res. 1996, 56, 1367–1373. [Google Scholar]
- Bar, J.; Lukaschuk, N.; Zalcenstein, A.; Wilder, S.; Seger, R.; Oren, M. The PI3K inhibitor LY294002 prevents p53 induction by DNA damage and attenuates chemotherapy-induced apoptosis. Cell Death Differ. 2005, 12, 1578–1587. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Jia, M.-Y.; Fang, W.-Y.; Chen, X.-J.; Mu, L.-L.; Wang, Z.-Y.; Shen, Y.; Xiang, R.-F.; Wang, L.-N.; Wang, L.; et al. FLT3 inhibition upregulates HDAC8 via FOXO to inactivate p53 and promote maintenance of FLT3-ITD+ acute myeloid leukemia. Blood 2020, 135, 1472–1483. [Google Scholar] [CrossRef]
- Janku, F.; Hong, D.S.; Fu, S.; Piha-Paul, S.A.; Naing, A.; Falchook, G.S.; Tsimberidou, A.M.; Stepanek, V.M.; Moulder, S.L.; Lee, J.J.; et al. Assessing PIK3CA and PTEN in Early-Phase Trials with PI3K/AKT/mTOR Inhibitors. Cell Rep. 2014, 6, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Reck, M.; Krzakowski, M.; Chmielowska, E.; Sebastian, M.; Hadler, D.; Fox, T.; Wang, Q.; Greenberg, J.; Beckman, R.A.; von Pawel, J. A randomized, double-blind, placebo-controlled phase 2 study of tigatuzumab (CS-1008) in combination with carboplatin/paclitaxel in patients with chemotherapy-naïve metastatic/unresectable non-small cell lung cancer. Lung Cancer 2013, 82, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Michmerhuizen, N.L.; Leonard, E.; Matovina, C.; Harris, M.; Herbst, G.; Kulkarni, A.; Zhai, J.; Jiang, H.; Carey, T.E.; Brenner, J.C. Rationale for Using Irreversible Epidermal Growth Factor Receptor Inhibitors in Combination with Phosphatidylinositol 3-Kinase Inhibitors for Advanced Head and Neck Squamous Cell Carcinoma. Mol. Pharmacol. 2019, 95, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Soulières, D.; Faivre, S.; Mesía, R.; Remenár, É.; Li, S.-H.; Karpenko, A.; Dechaphunkul, A.; Ochsenreither, S.; Kiss, L.A.; Lin, J.-C.; et al. Buparlisib and paclitaxel in patients with platinum-pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL-1): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2017, 18, 323–335. [Google Scholar] [CrossRef]
- Brisson, R.J.; Kochanny, S.; Arshad, S.; Dekker, A.; DeSouza, J.A.; Saloura, V.; Vokes, E.E.; Seiwert, T.Y. A pilot study of the pan-class I PI3K inhibitor buparlisib in combination with cetuximab in patients with recurrent or metastatic head and neck cancer. Head Neck 2019, 41, 3842–3849. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.A.; Riaz, N.; Fury, M.G.; McBride, S.M.; Michel, L.; Lee, N.Y.; Sherman, E.J.; Baxi, S.S.; Haque, S.S.; Katabi, N.; et al. A Phase 1b Study of Cetuximab and BYL719 (Alpelisib) Concurrent with Intensity Modulated Radiation Therapy in Stage III-IVB Head and Neck Squamous Cell Carcinoma. Int. J. Radiat. Oncol. 2020, 106, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Chesebrough, J.W.; Cartlidge, S.A.; Ricketts, S.-A.; Incognito, L.; Veldman-Jones, M.; Blakey, D.C.; Tabrizi, M.; Jallal, B.; Trail, P.A.; et al. Dual IGF-I/II–Neutralizing Antibody MEDI-573 Potently Inhibits IGF Signaling and Tumor Growth. Cancer Res. 2011, 71, 1029–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedbichler, K.; Hofmann, M.H.; Kroez, M.; Ostermann, E.; Lamche, H.R.; Koessl, C.; Borges, E.; Pollak, M.N.; Adolf, G.; Adam, P.J. Pharmacodynamic and Antineoplastic Activity of BI 836845, a Fully Human IGF Ligand-Neutralizing Antibody, and Mechanistic Rationale for Combination with Rapamycin. Mol. Cancer Ther. 2014, 13, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mireuta, M.; Birman, E.; Barmash, M.; Pollak, M. Quantification of Binding of IGF-1 to BI 836845, a Candidate Therapeutic Antibody Against IGF-1 and IGF-2, and Effects of This Antibody on IGF-1:IGFBP-3 Complexes In Vitro and in Male C57BL/6 Mice. Endocrinology 2014, 155, 703–715. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Rayes, R.F.; Elahi, S.M.; Lu, Y.; Hancock, M.A.; Massie, B.; Rowe, G.E.; Aomari, H.; Hossain, S.; Durocher, Y.; et al. The IGF-Trap: Novel Inhibitor of Carcinoma Growth and Metastasis. Mol. Cancer Ther. 2015, 14, 982–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frago, S.; Nicholls, R.D.; Strickland, M.; Hughes, J.; Williams, C.; Garner, L.; Surakhy, M.; Maclean, R.; Rezgui, D.; Prince, S.N.; et al. Functional evolution of IGF2:IGF2R domain 11 binding generates novel structural interactions and a specific IGF2 antagonist. Proc. Natl. Acad. Sci. USA 2016, 113, E2766–E2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, S.N.; Foulstone, E.J.; Zaccheo, O.J.; Williams, C.; Hassan, A.B. Functional evaluation of novel soluble insulin-like growth factor (IGF)-II–specific ligand traps based on modified domain 11 of the human IGF2 receptor. Mol. Cancer Ther. 2007, 6, 607–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaniotis, G.; Moffett, S.; Sulea, T.; Wang, N.; Elahi, S.M.; Lessard, E.; Baardsnes, J.; Perrino, S.; Durocher, Y.; Frystyk, J.; et al. Enhanced anti-metastatic bioactivity of an IGF-TRAP re-engineered to improve physicochemical properties. Sci. Rep. 2018, 8, 17361. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badarni, M.; Prasad, M.; Golden, A.; Bhattacharya, B.; Levin, L.; Yegodayev, K.M.; Dimitstein, O.; Joshua, B.-Z.; Cohen, L.; Khrameeva, E.; et al. IGF2 Mediates Resistance to Isoform-Selective-Inhibitors of the PI3K in HPV Positive Head and Neck Cancer. Cancers 2021, 13, 2250. https://doi.org/10.3390/cancers13092250
Badarni M, Prasad M, Golden A, Bhattacharya B, Levin L, Yegodayev KM, Dimitstein O, Joshua B-Z, Cohen L, Khrameeva E, et al. IGF2 Mediates Resistance to Isoform-Selective-Inhibitors of the PI3K in HPV Positive Head and Neck Cancer. Cancers. 2021; 13(9):2250. https://doi.org/10.3390/cancers13092250
Chicago/Turabian StyleBadarni, Mai, Manu Prasad, Artemiy Golden, Baisali Bhattacharya, Liron Levin, Ksenia M. Yegodayev, Orr Dimitstein, Ben-Zion Joshua, Limor Cohen, Ekaterina Khrameeva, and et al. 2021. "IGF2 Mediates Resistance to Isoform-Selective-Inhibitors of the PI3K in HPV Positive Head and Neck Cancer" Cancers 13, no. 9: 2250. https://doi.org/10.3390/cancers13092250
APA StyleBadarni, M., Prasad, M., Golden, A., Bhattacharya, B., Levin, L., Yegodayev, K. M., Dimitstein, O., Joshua, B.-Z., Cohen, L., Khrameeva, E., Kong, D., Porgador, A., Braiman, A., Grandis, J. R., Rotblat, B., & Elkabets, M. (2021). IGF2 Mediates Resistance to Isoform-Selective-Inhibitors of the PI3K in HPV Positive Head and Neck Cancer. Cancers, 13(9), 2250. https://doi.org/10.3390/cancers13092250