
Neuroendocrine Factors in Melanoma Pathogenesis

 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Melanoma and Stress
3. Neurotransmitters and Melanoma
3.1. Catecholamines
3.2. Glutamate
3.3. Serotonin
3.4. Cannabinoids
4. Neurohormones and Melanoma
4.1. The Corticotropin-Releasing Hormone–Proopiomelanocortin Axis
4.2. Alpha-MSH
4.3. Thyrotropin-Releasing Hormone
4.4. Somatostatin
4.5. Vasopressin
5. Neuropeptides and Melanoma
5.1. Substance P
5.2. Calcitonin Gene-Related Peptide
5.3. Bradykinin
5.4. Neuropeptide Y
5.5. Galanin
5.6. Gastrin-Releasing Peptide
5.7. Enkephalin
5.8. Beta-Endorphin
5.9. Vasoactive Intestinal Peptide
6. Cellular and Molecular Neuro-Immune Interactions in Melanoma
6.1. Mast Cells
6.2. Nitric Oxide
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Nikolaou, V.; Stratigos, A.J. Emerging trends in the epidemiology of melanoma. Br. J. Derm. 2014, 170, 11–19. [Google Scholar] [CrossRef]
- Liu, Y.; Sheikh, M.S. Melanoma: Molecular Pathogenesis and Therapeutic Management. Mol. Cell. Pharmacol. 2014, 6, 228. [Google Scholar] [PubMed]
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Feigelson, H.S.; Powers, J.D.; Kumar, M.; Carroll, N.M.; Pathy, A.; Ritzwoller, D.P. Melanoma incidence, recurrence, and mortality in an integrated healthcare system: A retrospective cohort study. Cancer Med. 2019, 8, 4508–4516. [Google Scholar] [CrossRef]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinnya, S.; De’Ambrosis, B. Stress and melanoma: Increasing the evidence towards a causal basis. Arch. Dermatol. Res. Arch. Dermatol. Forsch. 2013, 305, 851–856. [Google Scholar] [CrossRef]
- Tuong, W.; Cheng, L.S.; Armstrong, A.W. Melanoma: Epidemiology, diagnosis, treatment, and outcomes. Dermatol. Clin. 2012, 30, 113–124. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Gabreanu, G.; Lupu, A.; Caruntu, C.; Boda, D.; Zurac, S. Inflammation markers in cutaneous melanoma-edgy biomarkers for prognosis. Discoveries 2015, 3, e38. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Zurac, S. Immune parameters in the prognosis and therapy monitoring of cutaneous melanoma patients: Experience, role, and limitations. BioMed Res. Int. 2013, 2013, 107940. [Google Scholar] [CrossRef] [Green Version]
- Neagu, M.; Constantin, C.; Martin, D.; Albulescu, L.; Iacob, N.; Ighigeanu, D. Whole body microwave irradiation for improved dacarbazine therapeutical action in cutaneous melanoma mouse model. Radiol. Res. Pract. 2013, 2013, 414816. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.; Constantin, C.; Tanase, C. Immune-related biomarkers for diagnosis/prognosis and therapy monitoring of cutaneous melanoma. Expert Rev. Mol. Diagn. 2010, 10, 897–919. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Manda, G.; Margaritescu, I. Biomarkers of metastatic melanoma. Biomark. Med. 2009, 3, 71–89. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Caruntu, C.; Dumitru, C.; Surcel, M.; Zurac, S. Inflammation: A key process in skin tumorigenesis. Oncol. Lett. 2019, 17, 4068–4084. [Google Scholar] [CrossRef] [Green Version]
- Surcel, M.; Constantin, C.; Caruntu, C.; Zurac, S.; Neagu, M. Inflammatory Cytokine Pattern Is Sex-Dependent in Mouse Cutaneous Melanoma Experimental Model. J. Immunol. Res. 2017, 2017, 9212134. [Google Scholar] [CrossRef]
- Caruntu, C.; Mirica, A.; Roşca, A.E.; Mirica, R.; Caruntu, A.; Tampa, M.; Matei, C.; Constantin, C.; Neagu, M.; Badarau, A.I.; et al. The Role of Estrogens and Estrogen Receptors in Melanoma Development and Progression. Acta Endocrinol. 2016, 12, 234–241. [Google Scholar] [CrossRef]
- Colucci, R.; Moretti, S. The role of stress and beta-adrenergic system in melanoma: Current knowledge and possible therapeutic options. J. Cancer Res. Clin. Oncol. 2016, 142, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Caruntu, C.; Boda, D.; Constantin, C.; Caruntu, A.; Neagu, M. Catecholamines increase in vitro proliferation of murine B16F10 melanoma cells. Acta Endocrinol. 2014, 10, 545–558. [Google Scholar] [CrossRef] [Green Version]
- Tampa, M.; Matei, C.; Caruntu, C.; Poteca, T.; Mihaila, D.; Paunescu, C.; Pitigoi, G.; Georgescu, S.R.; Constantin, C.; Neagu, M. Cellular impedance measurement–Novel method for in vitro investigation of drug efficacy. Farmacia 2016, 64, 430–434. [Google Scholar]
- Roosterman, D.; Goerge, T.; Schneider, S.W.; Bunnett, N.W.; Steinhoff, M. Neuronal control of skin function: The skin as a neuroimmunoendocrine organ. Physiol. Rev. 2006, 86, 1309–1379. [Google Scholar] [CrossRef]
- Lupu, M.; Caruntu, A.; Caruntu, C.; Papagheorghe, L.M.L.; Ilie, M.A.; Voiculescu, V.; Boda, D.; Constantin, C.; Tanase, C.; Sifaki, M.; et al. Neuroendocrine factors: The missing link in nonmelanoma skin cancer (Review). Oncol. Rep. 2017, 38, 1327–1340. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, S.R.; Sarbu, M.I.; Matei, C.; Ilie, M.A.; Caruntu, C.; Constantin, C.; Neagu, M.; Tampa, M. Capsaicin: Friend or Foe in Skin Cancer and Other Related Malignancies? Nutrients 2017, 9, 1365. [Google Scholar] [CrossRef] [Green Version]
- Filippi, A.; Caruntu, C.; Gheorghe, R.O.; Deftu, A.; Amuzescu, B.; Ristoiu, V. Catecholamines reduce transient receptor potential vanilloid type 1 desensitization in cultured dorsal root ganglia neurons. J. Physiol. Pharmacol. 2016, 67, 843–850. [Google Scholar]
- Ginghina, O.; Negrei, C.; Hudita, A.; Lavric, V.; Galateanu, B.; Dragomir, S.; Burcea-Dragomiroiu, G.; Bârcă, M.; Nițipir, C.; Diaconu, C.; et al. In Vitro impact of some natural compounds on HT-29 colorectal adenocarcinoma cells. Farmacia 2017, 65, 947–953. [Google Scholar]
- Caruntu, C.; Boda, D.; Musat, S.; Caruntu, A.; Poenaru, E.; Calenic, B.; Savulescu-Fiedler, I.; Draghia, A.; Rotaru, M.; Badarau, A. Stress Effects on Cutaneous Nociceptive Nerve Fibers and Their Neurons of Origin in Rats. Rom. Biotechnol. Lett. 2014, 19, 9517–9530. [Google Scholar]
- Caruntu, C.; Grigore, C.; Caruntu, A.; Diaconeasa, A.; Boda, D. The role of stress in skin disease. Intern. Med. 2011, 8, 73–84. [Google Scholar]
- Caruntu, C.; Ilie Ghita, M.; Caruntu, A.; Boda, D. The role of stress in the multifactorial etiopathogenesis of acne. Rom. Med. J. 2011, 58, 98–101. [Google Scholar]
- Gupta, M.A.; Gupta, A.K. Psychiatric and psychological co-morbidity in patients with dermatologic disorders: Epidemiology and management. Am. J. Clin. Dermatol. 2003, 4, 833–842. [Google Scholar] [CrossRef]
- Sanzo, M.; Colucci, R.; Arunachalam, M.; Berti, S.; Moretti, S. Stress as a possible mechanism in melanoma progression. Derm. Res. Pract. 2010, 2010, 483493. [Google Scholar] [CrossRef] [PubMed]
- Fawzy, F.I.; Fawzy, N.W.; Hyun, C.S.; Elashoff, R.; Guthrie, D.; Fahey, J.L.; Morton, D.L. Malignant melanoma. Effects of an early structured psychiatric intervention, coping, and affective state on recurrence and survival 6 years later. Arch. Gen. Psychiatry 1993, 50, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Fawzy, F.I.; Canada, A.L.; Fawzy, N.W. Malignant melanoma: Effects of a brief, structured psychiatric intervention on survival and recurrence at 10-year follow-up. Arch. Gen. Psychiatry 2003, 60, 100–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temoshok, L.; Heller, B.W.; Sagebiel, R.W.; Blois, M.S.; Sweet, D.M.; DiClemente, R.J.; Gold, M.L. The relationship of psychosocial factors to prognostic indicators in cutaneous malignant melanoma. J. Psychosom. Res. 1985, 29, 139–153. [Google Scholar] [CrossRef]
- Temoshok, L. Biopsychosocial studies on cutaneous malignant melanoma: Psychosocial factors associated with prognostic indicators, progression, psychophysiology and tumor-host response. Soc. Sci. Med. 1985, 20, 833–840. [Google Scholar] [CrossRef]
- Beswick, S.; Affleck, P.; Elliott, F.; Gerry, E.; Boon, A.; Bale, L.; Nolan, C.; Barrett, J.H.; Bertram, C.; Marsden, J.; et al. Environmental risk factors for relapse of melanoma. Eur. J. Cancer 2008, 44, 1717–1725. [Google Scholar] [CrossRef] [Green Version]
- Lehto, U.S.; Ojanen, M.; Dyba, T.; Aromaa, A.; Kellokumpu-Lehtinen, P. Baseline psychosocial predictors of survival in localized melanoma. J. Psychosom. Res. 2007, 63, 9–15. [Google Scholar] [CrossRef]
- Butow, P.N.; Coates, A.S.; Dunn, S.M. Psychosocial predictors of survival in metastatic melanoma. J. Clin. Oncol. 1999, 17, 2256–2263. [Google Scholar] [CrossRef]
- Rogentine, G.N., Jr.; van Kammen, D.P.; Fox, B.H.; Docherty, J.P.; Rosenblatt, J.E.; Boyd, S.C.; Bunney, W.E., Jr. Psychological factors in the prognosis of malignant melanoma: A prospective study. Psychosom. Med. 1979, 41, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, J.; Reisine, T.D. Stress hormones: Their interaction and regulation. Science 1984, 224, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.V.; Kim, S.J.; Donovan, E.L.; Chen, M.; Gross, A.C.; Webster Marketon, J.I.; Barsky, S.H.; Glaser, R. Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: Implications for stress-related enhancement of tumor progression. Brain Behav. Immun. 2009, 23, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Kanno, J.; Wakikawa, A.; Utsuyama, M.; Hirokawa, K. Effect of restraint stress on immune system and experimental B16 melanoma metastasis in aged mice. Mech. Ageing Dev. 1997, 93, 107–117. [Google Scholar] [CrossRef]
- Higashiyama, A.; Watanabe, H.; Okumura, K.; Yagita, H. Involvement of tumor necrosis factor alpha and very late activation antigen 4/vascular cell adhesion molecule 1 interaction in surgical-stress-enhanced experimental metastasis. Cancer Immunol. Immunother. 1996, 42, 231–236. [Google Scholar] [CrossRef]
- Azpiroz, A.; De Miguel, Z.; Fano, E.; Vegas, O. Relations between different coping strategies for social stress, tumor development and neuroendocrine and immune activity in male mice. Brain Behav. Immun. 2008, 22, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.; Kiecolt-Glaser, J.K. Stress-induced immune dysfunction: Implications for health. Nat. Rev. Immunol. 2005, 5, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.D.; Campisi, J.; Sharkey, C.M.; Kennedy, S.L.; Nickerson, M.; Greenwood, B.N.; Fleshner, M. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience 2005, 135, 1295–1307. [Google Scholar] [CrossRef]
- Kiecolt-Glaser, J.K.; Loving, T.J.; Stowell, J.R.; Malarkey, W.B.; Lemeshow, S.; Dickinson, S.L.; Glaser, R. Hostile marital interactions, proinflammatory cytokine production, and wound healing. Arch. Gen. Psychiatry 2005, 62, 1377–1384. [Google Scholar] [CrossRef]
- Segerstrom, S.C.; Miller, G.E. Psychological stress and the human immune system: A meta-analytic study of 30 years of inquiry. Psychol. Bull. 2004, 130, 601–630. [Google Scholar] [CrossRef] [Green Version]
- Padgett, D.A.; Glaser, R. How stress influences the immune response. Trends Immunol. 2003, 24, 444–448. [Google Scholar] [CrossRef]
- Valles, S.L.; Benlloch, M.; Rodriguez, M.L.; Mena, S.; Pellicer, J.A.; Asensi, M.; Obrador, E.; Estrela, J.M. Stress hormones promote growth of B16-F10 melanoma metastases: An interleukin 6- and glutathione-dependent mechanism. J. Transl. Med. 2013, 11, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, E.V. Role for catecholamines in tumor progression: Possible use for beta-blockers in the treatment of cancer. Cancer Biol. Ther. 2010, 10, 30–32. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shaanan, T.L.; Schiller, M.; Azulay-Debby, H.; Korin, B.; Boshnak, N.; Koren, T.; Krot, M.; Shakya, J.; Rahat, M.A.; Hakim, F.; et al. Modulation of anti-tumor immunity by the brain’s reward system. Nat. Commun. 2018, 9, 2723. [Google Scholar] [CrossRef] [Green Version]
- Pope, S.; Heales, S.J. Neurotransmitters; Elsevier: Amsterdam, The Netherlands, 2018; p. 14. [Google Scholar]
- Horvathova, L.; Padova, A.; Tillinger, A.; Osacka, J.; Bizik, J.; Mravec, B. Sympathectomy reduces tumor weight and affects expression of tumor-related genes in melanoma tissue in the mouse. Stress 2016, 19, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Petsko, G.A.; Eliezer, D. Parkinson’s Disease and Melanoma: Co-Occurrence and Mechanisms. J. Parkinson’s Dis. 2018, 8, 385–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalvin, L.A.; Damento, G.M.; Yawn, B.P.; Abbott, B.A.; Hodge, D.O.; Pulido, J.S. Parkinson Disease and Melanoma: Confirming and Reexamining an Association. Mayo Clin. Proc. 2017, 92, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Dube, U.; Ibanez, L.; Budde, J.P.; Benitez, B.A.; Davis, A.A.; Harari, O.; Iles, M.M.; Law, M.H.; Brown, K.M.; Cruchaga, C. Overlapping genetic architecture between Parkinson disease and melanoma. Acta Neuropathol. 2020, 139, 347–364. [Google Scholar] [CrossRef]
- Tang, J.; Li, Z.; Lu, L.; Cho, C.H. beta-Adrenergic system, a backstage manipulator regulating tumour progression and drug target in cancer therapy. Semin. Cancer Biol. 2013, 23, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarparo, A.C.; Sumida, D.H.; Patrao, M.T.; Avellar, M.C.; Visconti, M.A.; Maria de Lauro Castrucci, A. Catecholamine effects on human melanoma cells evoked by alpha1-adrenoceptors. Arch. Dermatol. Res. 2004, 296, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Scarparo, A.C.; Visconti, M.A.; de Oliveira, A.R.; Castrucci, A.M. Adrenoceptors in normal and malignant human melanocytes. Arch. Dermatol. Res. 2000, 292, 265–267. [Google Scholar] [CrossRef]
- Moretti, S.; Massi, D.; Farini, V.; Baroni, G.; Parri, M.; Innocenti, S.; Cecchi, R.; Chiarugi, P. beta-adrenoceptors are upregulated in human melanoma and their activation releases pro-tumorigenic cytokines and metalloproteases in melanoma cell lines. Lab. Investig. 2013, 93, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Janik, M.E.; Szlezak, D.; Surman, M.; Golas, A.; Litynska, A.; Przybylo, M. Diversified beta-2-adrenergic Receptor Expression and Action in Melanoma Cells. Anticancer Res. 2017, 37, 3025–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef] [Green Version]
- Moretti, S.; Pinzi, C.; Spallanzani, A.; Berti, E.; Chiarugi, A.; Mazzoli, S.; Fabiani, M.; Vallecchi, C.; Herlyn, M. Immunohistochemical evidence of cytokine networks during progression of human melanocytic lesions. Int. J. Cancer 1999, 84, 160–168. [Google Scholar] [CrossRef]
- Mahabeleshwar, G.H.; Byzova, T.V. Angiogenesis in melanoma. Semin. Oncol. 2007, 34, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Loffek, S.; Zigrino, P.; Steiger, J.; Kurschat, P.; Smola, H.; Mauch, C. Melanoma cell-derived vascular endothelial growth factor induces endothelial tubulogenesis within fibrin gels by a metalloproteinase-mediated mechanism. Eur. J. Cell Biol. 2006, 85, 1167–1177. [Google Scholar] [CrossRef]
- Osella-Abate, S.; Quaglino, P.; Savoia, P.; Leporati, C.; Comessatti, A.; Bernengo, M.G. VEGF-165 serum levels and tyrosinase expression in melanoma patients: Correlation with the clinical course. Melanoma Res. 2002, 12, 325–334. [Google Scholar] [CrossRef]
- Bar-Eli, M. Role of interleukin-8 in tumor growth and metastasis of human melanoma. Pathobiology 1999, 67, 12–18. [Google Scholar] [CrossRef]
- Payne, A.S.; Cornelius, L.A. The role of chemokines in melanoma tumor growth and metastasis. J. Investig. Dermatol. 2002, 118, 915–922. [Google Scholar] [CrossRef] [Green Version]
- Ene, C.D.; Tampa, M.; Nicolae, I.; Mitran, C.I.; Mitran, M.I.; Matei, C.; Caruntu, A.; Caruntu, C.; Georgescu, S.R. Antiganglioside Antibodies and Inflammatory Response in Cutaneous Melanoma. J. Immunol. Res. 2020, 2020, 2491265. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, A.; Kaira, K.; Mori, K.; Kato, M.; Shimizu, K.; Yasuda, M.; Takahashi, A.; Oyama, T.; Asao, T.; Ishikawa, O. Prognostic significance of beta2-adrenergic receptor expression in malignant melanoma. Tumour Biol. 2016, 37, 5971–5978. [Google Scholar] [CrossRef]
- Dal Monte, M.; Casini, G.; Filippi, L.; Nicchia, G.P.; Svelto, M.; Bagnoli, P. Functional involvement of beta3-adrenergic receptors in melanoma growth and vascularization. J. Mol. Med. 2013, 91, 1407–1419. [Google Scholar] [CrossRef]
- Filippi, L.; Bruno, G.; Domazetovic, V.; Favre, C.; Calvani, M. Current Therapies and New Targets to Fight Melanoma: A Promising Role for the β3-Adrenoreceptor. Cancers 2020, 12, 1415. [Google Scholar] [CrossRef]
- Calvani, M.; Pelon, F.; Comito, G.; Taddei, M.L.; Moretti, S.; Innocenti, S.; Nassini, R.; Gerlini, G.; Borgognoni, L.; Bambi, F.; et al. Norepinephrine promotes tumor microenvironment reactivity through beta3-adrenoreceptors during melanoma progression. Oncotarget 2015, 6, 4615–4632. [Google Scholar] [CrossRef] [Green Version]
- Calvani, M.; Bruno, G.; Dabraio, A.; Subbiani, A.; Bianchini, F.; Fontani, F.; Casazza, G.; Vignoli, M.; De Logu, F.; Frenos, S.; et al. β3-Adrenoreceptor Blockade Induces Stem Cells Differentiation in Melanoma Microenvironment. Int. J. Mol. Sci. 2020, 21, 1420. [Google Scholar] [CrossRef] [Green Version]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Kharaishvili, G.; Simkova, D.; Bouchalova, K.; Gachechiladze, M.; Narsia, N.; Bouchal, J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell Int. 2014, 14, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, E.V.; Eubank, T.D. The impact of adrenergic signaling in skin cancer progression: Possible repurposing of beta-blockers for treatment of skin cancer. Cancer Biomark. 2013, 13, 155–160. [Google Scholar] [CrossRef]
- Tlsty, T.D.; Coussens, L.M. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. 2006, 1, 119–150. [Google Scholar] [CrossRef]
- Calvani, M.; Bruno, G.; Dal Monte, M.; Nassini, R.; Fontani, F.; Casini, A.; Cavallini, L.; Becatti, M.; Bianchini, F.; De Logu, F.; et al. β(3) -Adrenoceptor as a potential immuno-suppressor agent in melanoma. Br. J. Pharmacol. 2019, 176, 2509–2524. [Google Scholar] [CrossRef]
- Magnoni, C.; Giudice, S.; Pellacani, G.; Bertazzoni, G.; Longo, C.; Veratti, E.; Morini, D.; Benassi, L.; Vaschieri, C.; Azzoni, P.; et al. Stem cell properties in cell cultures from different stage of melanoma progression. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 171–181. [Google Scholar] [CrossRef]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef] [Green Version]
- Nguyễn, L.T.H. The roles of beta-adrenergic receptors in tumorigenesis and the possible use of beta-adrenergic blockers for cancer treatment: Possible genetic and cell-signaling mechanisms. Cancer Manag. Res. 2012, 4, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Lemeshow, S.; Sorensen, H.T.; Phillips, G.; Yang, E.V.; Antonsen, S.; Riis, A.H.; Lesinski, G.B.; Jackson, R.; Glaser, R. beta-Blockers and survival among Danish patients with malignant melanoma: A population-based cohort study. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2273–2279. [Google Scholar] [CrossRef] [Green Version]
- De Giorgi, V.; Grazzini, M.; Gandini, S.; Benemei, S.; Lotti, T.; Marchionni, N.; Geppetti, P. Treatment with beta-blockers and reduced disease progression in patients with thick melanoma. Arch. Intern. Med. 2011, 171, 779–781. [Google Scholar] [CrossRef]
- De Giorgi, V.; Gandini, S.; Grazzini, M.; Benemei, S.; Marchionni, N.; Geppetti, P. Effect of beta-blockers and other antihypertensive drugs on the risk of melanoma recurrence and death. Mayo Clin. Proc. 2013, 88, 1196–1203. [Google Scholar] [CrossRef]
- Kokolus, K.M.; Zhang, Y.; Sivik, J.M.; Schmeck, C.; Zhu, J.; Repasky, E.A.; Drabick, J.J.; Schell, T.D. Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology 2018, 7, e1405205. [Google Scholar] [CrossRef] [Green Version]
- Sereni, F.; Dal Monte, M.; Filippi, L.; Bagnoli, P. Role of host beta1- and beta2-adrenergic receptors in a murine model of B16 melanoma: Functional involvement of beta3-adrenergic receptors. Naunyn Schmiedebergs Arch. Pharmacol. 2015, 388, 1317–1331. [Google Scholar] [CrossRef]
- Dal Monte, M.; Fornaciari, I.; Nicchia, G.P.; Svelto, M.; Casini, G.; Bagnoli, P. beta3-adrenergic receptor activity modulates melanoma cell proliferation and survival through nitric oxide signaling. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 533–543. [Google Scholar] [CrossRef]
- Surcel, M.; Caruntu, C.; Tampa, M.; Matei, C.; PiȚUru, S.; Georgescu, S.R.; Constantin, C.; Zurac, S.; Neagu, M. Adrenergic modulation of melanoma cells proliferation. Farmacia 2018, 66, 820–825. [Google Scholar] [CrossRef]
- McCourt, C.; Coleman, H.G.; Murray, L.J.; Cantwell, M.M.; Dolan, O.; Powe, D.G.; Cardwell, C.R. Beta-blocker usage after malignant melanoma diagnosis and survival: A population-based nested case-control study. Br. J. Dermatol. 2014, 170, 930–938. [Google Scholar] [CrossRef]
- Livingstone, E.; Hollestein, L.M.; van Herk-Sukel, M.P.; van de Poll-Franse, L.; Nijsten, T.; Schadendorf, D.; de Vries, E. beta-Blocker use and all-cause mortality of melanoma patients: Results from a population-based Dutch cohort study. Eur. J. Cancer 2013, 49, 3863–3871. [Google Scholar] [CrossRef]
- Williams, N.M.; Vincent, L.T.; Rodriguez, G.A.; Nouri, K. Antihypertensives and melanoma: An updated review. Pigment Cell Melanoma Res 2020, 33, 806–813. [Google Scholar] [CrossRef]
- Jean Wrobel, L.; Bod, L.; Lengagne, R.; Kato, M.; Prevost-Blondel, A.; Le Gal, F.A. Propranolol induces a favourable shift of anti-tumor immunity in a murine spontaneous model of melanoma. Oncotarget 2016, 7, 77825–77837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Monte, M.; Calvani, M.; Cammalleri, M.; Favre, C.; Filippi, L.; Bagnoli, P. β-Adrenoceptors as drug targets in melanoma: Novel preclinical evidence for a role of β(3) -adrenoceptors. Br. J. Pharmacol. 2019, 176, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.; Luu, B. Can propranolol prevent progression of melanoma? Jaapa 2019, 32, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.; Idorn, M.; Olofsson, G.H.; Lauenborg, B.; Nookaew, I.; Hansen, R.H.; Johannesen, H.H.; Becker, J.C.; Pedersen, K.S.; Dethlefsen, C.; et al. Voluntary Running Suppresses Tumor Growth through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution. Cell Metab. 2016, 23, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarparo, A.C.; Visconti, M.A.; Castrucci, A.M. Signalling pathways evoked by alpha1-adrenoceptors in human melanoma cells. Cell Biochem. Funct. 2006, 24, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Mangia, S.; Giove, F.; Dinuzzo, M. Metabolic pathways and activity-dependent modulation of glutamate concentration in the human brain. Neurochem. Res. 2012, 37, 2554–2561. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.Y.; Chang, K.; Pickel, J.M.; Badger, J.D., 2nd; Roche, K.W. Expression of the metabotropic glutamate receptor 5 (mGluR5) induces melanoma in transgenic mice. Proc. Natl. Acad. Sci. USA 2011, 108, 15219–15224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehnert, J.M.; Silk, A.W.; Lee, J.H.; Dudek, L.; Jeong, B.S.; Li, J.; Schenkel, J.M.; Sadimin, E.; Kane, M.; Lin, H.; et al. A phase II trial of riluzole, an antagonist of metabotropic glutamate receptor 1 (GRM1) signaling, in patients with advanced melanoma. Pigment Cell Melanoma Res. 2018, 31, 534–540. [Google Scholar] [CrossRef]
- Abdel-Daim, M.; Funasaka, Y.; Komoto, M.; Nakagawa, Y.; Yanagita, E.; Nishigori, C. Pharmacogenomics of metabotropic glutamate receptor subtype 1 and in vivo malignant melanoma formation. J. Dermatol. 2010, 37, 635–646. [Google Scholar] [CrossRef]
- Pollock, P.M.; Cohen-Solal, K.; Sood, R.; Namkoong, J.; Martino, J.J.; Koganti, A.; Zhu, H.; Robbins, C.; Makalowska, I.; Shin, S.S.; et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat. Genet. 2003, 34, 108–112. [Google Scholar] [CrossRef]
- Ohtani, Y.; Harada, T.; Funasaka, Y.; Nakao, K.; Takahara, C.; Abdel-Daim, M.; Sakai, N.; Saito, N.; Nishigori, C.; Aiba, A. Metabotropic glutamate receptor subtype-1 is essential for in vivo growth of melanoma. Oncogene 2008, 27, 7162–7170. [Google Scholar] [CrossRef] [Green Version]
- Namkoong, J.; Shin, S.S.; Lee, H.J.; Marin, Y.E.; Wall, B.A.; Goydos, J.S.; Chen, S. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007, 67, 2298–2305. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.; Singh, S.J.; Eddy, K.; Filipp, F.V.; Chen, S. Concurrent Targeting of Glutaminolysis and Metabotropic Glutamate Receptor 1 (GRM1) Reduces Glutamate Bioavailability in GRM1(+) Melanoma. Cancer Res. 2019, 79, 1799–1809. [Google Scholar] [CrossRef] [Green Version]
- D’Mello, S.A.; Joseph, W.R.; Green, T.N.; Leung, E.Y.; During, M.J.; Finlay, G.J.; Baguley, B.C.; Kalev-Zylinska, M.L. Selected GRIN2A mutations in melanoma cause oncogenic effects that can be modulated by extracellular glutamate. Cell Calcium 2016, 60, 384–395. [Google Scholar] [CrossRef]
- Gelb, T.; Pshenichkin, S.; Hathaway, H.A.; Grajkowska, E.; Dalley, C.B.; Wolfe, B.B.; Wroblewski, J.T. Atypical signaling of metabotropic glutamate receptor 1 in human melanoma cells. Biochem. Pharmacol. 2015, 98, 182–189. [Google Scholar] [CrossRef]
- Wangari-Talbot, J.; Goydos, J.; Chen, S. Role of the G Protein-Coupled Receptor, mGlu₁, in Melanoma Development. Pharmaceuticals 2010, 3, 2821–2837. [Google Scholar] [CrossRef] [PubMed]
- Isola, A.L.; Eddy, K.; Zembrzuski, K.; Goydos, J.S.; Chen, S. Exosomes released by metabotropic glutamate receptor 1 (GRM1) expressing melanoma cells increase cell migration and invasiveness. Oncotarget 2018, 9, 1187–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Li, J.; Koo, J.; Shin, S.S.; Lin, Y.; Jeong, B.S.; Mehnert, J.M.; Chen, S.; Cohen-Sola, K.A.; Goydos, J.S. Activation of the glutamate receptor GRM1 enhances angiogenic signaling to drive melanoma progression. Cancer Res. 2014, 74, 2499–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neto, A.; Ceol, C.J. Melanoma-associated GRM3 variants dysregulate melanosome trafficking and cAMP signaling. Pigment Cell Melanoma Res 2018, 31, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Rzeski, W.; Turski, L.; Ikonomidou, C. Glutamate antagonists limit tumor growth. Proc. Natl. Acad. Sci. USA 2001, 98, 6372–6377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; He, C.D.; Liu, J.; Sun, C.; Lu, P.; Li, L.; Gao, L.; Zhang, Y.; Xu, Y.; Shan, L.; et al. Blocking glutamate-mediated signalling inhibits human melanoma growth and migration. Exp. Dermatol. 2012, 21, 926–931. [Google Scholar] [CrossRef]
- Xie, L.; Hanyu, M.; Fujinaga, M.; Zhang, Y.; Hu, K.; Minegishi, K.; Jiang, C.; Kurosawa, F.; Morokoshi, Y.; Li, H.K.; et al. (131)I-IITM and (211)At-AITM: Two Novel Small-Molecule Radiopharmaceuticals Targeting Oncoprotein Metabotropic Glutamate Receptor 1. J. Nucl. Med. 2020, 61, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, N.; Tachibana, M.; Ago, Y.; Goda, H.; Sakurai, F.; Mizuguchi, H. LY341495, an mGluR2/3 Antagonist, Regulates the Immunosuppressive Function of Myeloid-Derived Suppressor Cells and Inhibits Melanoma Tumor Growth. Biol. Pharm. Bull. 2018, 41, 1866–1869. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, M.P.; Nunes-Correia, I.; Santos, A.E.; Custódio, J.B. The combination of glutamate receptor antagonist MK-801 with tamoxifen and its active metabolites potentiates their antiproliferative activity in mouse melanoma K1735-M2 cells. Exp. Cell Res. 2014, 321, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Yip, D.; Le, M.N.; Chan, J.L.; Lee, J.H.; Mehnert, J.A.; Yudd, A.; Kempf, J.; Shih, W.J.; Chen, S.; Goydos, J.S. A phase 0 trial of riluzole in patients with resectable stage III and IV melanoma. Clin. Cancer Res. 2009, 15, 3896–3902. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Wall, B.A.; Wangari-Talbot, J.; Shin, S.S.; Rosenberg, S.; Chan, J.L.; Namkoong, J.; Goydos, J.S.; Chen, S. Glutamatergic pathway targeting in melanoma: Single-agent and combinatorial therapies. Clin. Cancer Res. 2011, 17, 7080–7092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abushahba, W.; Olabisi, O.O.; Jeong, B.S.; Boregowda, R.K.; Wen, Y.; Liu, F.; Goydos, J.S.; Lasfar, A.; Cohen-Solal, K.A. Non-canonical Smads phosphorylation induced by the glutamate release inhibitor, riluzole, through GSK3 activation in melanoma. PLoS ONE 2012, 7, e47312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemieszek, M.K.; Stepulak, A.; Sawa-Wejksza, K.; Czerwonka, A.; Ikonomidou, C.; Rzeski, W. Riluzole Inhibits Proliferation, Migration and Cell Cycle Progression and Induces Apoptosis in Tumor Cells of Various Origins. Anti-Cancer Agents Med. Chem. 2018, 18, 565–572. [Google Scholar] [CrossRef]
- Pelletier, J.C.; Chen, S.; Bian, H.; Shah, R.; Smith, G.R.; Wrobel, J.E.; Reitz, A.B. Dipeptide Prodrugs of the Glutamate Modulator Riluzole. ACS Med. Chem. Lett. 2018, 9, 752–756. [Google Scholar] [CrossRef]
- Olivier, B. Serotonin: A never-ending story. Eur. J. Pharmacol. 2015, 753, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Geng, K.K.; Ping, F.F.; Gao, Y.Y.; Liu, L.; Feng, B.N. Cross-talk between 5-hydroxytryptamine and substance P in the melanogensis and apoptosis of B16F10 melanoma cells. Eur. J. Pharmacol. 2016, 775, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Park, M.K.; Kim, S.Y.; Park Choo, H.Y.; Lee, A.Y.; Lee, C.H. Serotonin induces melanogenesis via serotonin receptor 2A. Br. J. Dermatol. 2011, 165, 1344–1348. [Google Scholar] [CrossRef]
- Nordlind, K.; Azmitia, E.C.; Slominski, A. The skin as a mirror of the soul: Exploring the possible roles of serotonin. Exp. Dermatol. 2008, 17, 301–311. [Google Scholar] [CrossRef]
- Ptak, W.; Geba, G.P.; Askenase, P.W. Initiation of delayed-type hypersensitivity by low doses of monoclonal IgE antibody. Mediation by serotonin and inhibition by histamine. J. Immunol. 1991, 146, 3929–3936. [Google Scholar]
- Slominski, A.; Wortsman, J.; Tobin, D.J. The cutaneous serotoninergic/melatoninergic system: Securing a place under the sun. FASEB J. 2005, 19, 176–194. [Google Scholar] [CrossRef]
- Horai, T.; Nishihara, H.; Hattori, S.; Tateishi, R. Malignant melanoma producing serotonin. Cancer 1979, 43, 294–298. [Google Scholar] [CrossRef]
- Slominski, A.; Wortsman, J.; Kohn, L.; Ain, K.B.; Venkataraman, G.M.; Pisarchik, A.; Chung, J.H.; Giuliani, C.; Thornton, M.; Slugocki, G.; et al. Expression of hypothalamic-pituitary-thyroid axis related genes in the human skin. J. Investig. Dermatol. 2002, 119, 1449–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezes, A.C.; Carvalheiro, M.; Ferreira de Oliveira, J.M.P.; Ascenso, A.; Oliveira, H. Cytotoxic effect of the serotonergic drug 1-(1-Naphthyl)piperazine against melanoma cells. Toxicol. In Vitro 2018, 47, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Kawano, Y.; Yamanaka, A.; Maruyama, S. N-[(Dihydroxyphenyl)acyl]serotonins as potent inhibitors of tyrosinase from mouse and human melanoma cells. Bioorg. Med. Chem. Lett. 2009, 19, 4178–4182. [Google Scholar] [CrossRef] [PubMed]
- Naimi-Akbar, C.; Ritter, M.; Demel, S.; El-Nour, H.; Hedblad, M.A.; Azmitia, E.C.; Nordlind, K. Different serotonergic expression in nevomelanocytic tumors. Cancers 2010, 2, 1166–1177. [Google Scholar] [CrossRef]
- Peters, M.A.M.; Meijer, C.; Fehrmann, R.S.N.; Walenkamp, A.M.E.; Kema, I.P.; de Vries, E.G.E.; Hollema, H.; Oosting, S.F. Serotonin and Dopamine Receptor Expression in Solid Tumours Including Rare Cancers. Pathol. Oncol. Res. 2020, 26, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Stachura, P.; Xu, H.C.; Umesh Ganesh, N.; Cox, F.; Wang, R.; Lang, K.S.; Gopalakrishnan, J.; Häussinger, D.; Homey, B.; et al. Repurposing the serotonin agonist Tegaserod as an anticancer agent in melanoma: Molecular mechanisms and clinical implications. J. Exp. Clin. Cancer Res. 2020, 39, 38. [Google Scholar] [CrossRef] [Green Version]
- Lebena, A.; Vegas, O.; Gomez-Lazaro, E.; Arregi, A.; Garmendia, L.; Beitia, G.; Azpiroz, A. Melanoma tumors alter proinflammatory cytokine production and monoamine brain function, and induce depressive-like behavior in male mice. Behav. Brain Res. 2014, 272, 83–92. [Google Scholar] [CrossRef]
- Grygier, B.; Arteta, B.; Kubera, M.; Basta-Kaim, A.; Budziszewska, B.; Leskiewicz, M.; Curzytek, K.; Duda, W.; Lason, W.; Maes, M. Inhibitory effect of antidepressants on B16F10 melanoma tumor growth. Pharmacol. Rep. 2013, 65, 672–681. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Fu, M.; Pei, S.; Zhou, L.; Shang, J. R-Fluoxetine Increases Melanin Synthesis Through a 5-HT1A/2A Receptor and p38 MAPK Signaling Pathways. Int. J. Mol. Sci. 2018, 20, 80. [Google Scholar] [CrossRef] [Green Version]
- Boia-Ferreira, M.; Basílio, A.B.; Hamasaki, A.E.; Matsubara, F.H.; Appel, M.H.; Da Costa, C.R.V.; Amson, R.; Telerman, A.; Chaim, O.M.; Veiga, S.S.; et al. TCTP as a therapeutic target in melanoma treatment. Br. J. Cancer 2017, 117, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Kubera, M.; Grygier, B.; Arteta, B.; Urbanska, K.; Basta-Kaim, A.; Budziszewska, B.; Leskiewicz, M.; Kolaczkowska, E.; Maes, M.; Szczepanik, M.; et al. Age-dependent stimulatory effect of desipramine and fluoxetine pretreatment on metastasis formation by B16F10 melanoma in male C57BL/6 mice. Pharmacol. Rep. 2009, 61, 1113–1126. [Google Scholar] [CrossRef] [Green Version]
- Berge, L.A.M.; Andreassen, B.K.; Stenehjem, J.S.; Heir, T.; Furu, K.; Juzeniene, A.; Roscher, I.; Larsen, I.K.; Green, A.C.; Veierød, M.B.; et al. Use of Antidepressants and Risk of Cutaneous Melanoma: A Prospective Registry-Based Case-Control Study. Clin. Epidemiol. 2020, 12, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boursi, B.; Lurie, I.; Haynes, K.; Mamtani, R.; Yang, Y.X. Chronic therapy with selective serotonin reuptake inhibitors and survival in newly diagnosed cancer patients. Eur. J. Cancer Care 2018, 27. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.M.; Fowler, C.J. The endocannabinoid system: Drug targets, lead compounds, and potential therapeutic applications. J. Med. Chem. 2005, 48, 5059–5087. [Google Scholar] [CrossRef]
- Adinolfi, B.; Romanini, A.; Vanni, A.; Martinotti, E.; Chicca, A.; Fogli, S.; Nieri, P. Anticancer activity of anandamide in human cutaneous melanoma cells. Eur. J. Pharmacol. 2013, 718, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Scheau, C.; Badarau, I.A.; Mihai, L.-G.; Scheau, A.-E.; Costache, D.O.; Constantin, C.; Calina, D.; Caruntu, C.; Costache, R.S.; Caruntu, A. Cannabinoids in the Pathophysiology of Skin Inflammation. Molecules 2020, 25, 652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazquez, C.; Carracedo, A.; Barrado, L.; Real, P.J.; Fernandez-Luna, J.L.; Velasco, G.; Malumbres, M.; Guzman, M. Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J. 2006, 20, 2633–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpi, S.; Fogli, S.; Polini, B.; Montagnani, V.; Podestà, A.; Breschi, M.C.; Romanini, A.; Stecca, B.; Nieri, P. Tumor-promoting effects of cannabinoid receptor type 1 in human melanoma cells. Toxicol. In Vitro 2017, 40, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Kenessey, I.; Banki, B.; Mark, A.; Varga, N.; Tovari, J.; Ladanyi, A.; Raso, E.; Timar, J. Revisiting CB1 receptor as drug target in human melanoma. Pathol. Oncol. Res. 2012, 18, 857–866. [Google Scholar] [CrossRef]
- Sarnataro, D.; Pisanti, S.; Santoro, A.; Gazzerro, P.; Malfitano, A.M.; Laezza, C.; Bifulco, M. The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits human breast cancer cell proliferation through a lipid raft-mediated mechanism. Mol. Pharmacol. 2006, 70, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Simmerman, E.; Qin, X.; Yu, J.C.; Baban, B. Cannabinoids as a Potential New and Novel Treatment for Melanoma: A Pilot Study in a Murine Model. J. Surg. Res. 2019, 235, 210–215. [Google Scholar] [CrossRef]
- Scuderi, M.R.; Cantarella, G.; Scollo, M.; Lempereur, L.; Palumbo, M.; Saccani-Jotti, G.; Bernardini, R. The antimitogenic effect of the cannabinoid receptor agonist WIN55212-2 on human melanoma cells is mediated by the membrane lipid raft. Cancer Lett. 2011, 310, 240–249. [Google Scholar] [CrossRef]
- Armstrong, J.L.; Hill, D.S.; McKee, C.S.; Hernandez-Tiedra, S.; Lorente, M.; Lopez-Valero, I.; Eleni Anagnostou, M.; Babatunde, F.; Corazzari, M.; Redfern, C.P.F.; et al. Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J. Investig. Dermatol. 2015, 135, 1629–1637. [Google Scholar] [CrossRef] [Green Version]
- Bachari, A.; Piva, T.J.; Salami, S.A.; Jamshidi, N.; Mantri, N. Roles of Cannabinoids in Melanoma: Evidence from In Vivo Studies. Int. J. Mol. Sci. 2020, 21, 6040. [Google Scholar] [CrossRef]
- Taha, T.; Meiri, D.; Talhamy, S.; Wollner, M.; Peer, A.; Bar-Sela, G. Cannabis Impacts Tumor Response Rate to Nivolumab in Patients with Advanced Malignancies. Oncologist 2019, 24, 549–554. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.; Wortsman, J.; Luger, T.; Paus, R.; Solomon, S. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol. Rev. 2000, 80, 979–1020. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A. Neuroendocrine system of the skin. Dermatology 2005, 211, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slominski, A.; Wortsman, J. Neuroendocrinology of the skin. Endocr. Rev. 2000, 21, 457–487. [Google Scholar] [CrossRef]
- Kim, M.H.; Cho, D.; Kim, H.J.; Chong, S.J.; Lee, K.H.; Yu, D.S.; Park, C.J.; Lee, J.Y.; Cho, B.K.; Park, H.J. Investigation of the corticotropin-releasing hormone-proopiomelanocortin axis in various skin tumours. Br. J. Dermatol. 2006, 155, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Funasaka, Y.; Sato, H.; Chakraborty, A.K.; Ohashi, A.; Chrousos, G.P.; Ichihashi, M. Expression of proopiomelanocortin, corticotropin-releasing hormone (CRH), and CRH receptor in melanoma cells, nevus cells, and normal human melanocytes. J. Investig. Dermatol. Symp. Proc. 1999, 4, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Nagashima, Y.; Chrousos, G.P.; Ichihashi, M.; Funasak, Y. The expression of corticotropin-releasing hormone in melanoma. Pigment Cell Res. 2002, 15, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Park, H.; Yang, Y.; Kim, T.S.; Bang, S.I.; Cho, D. Enhancement of cell migration by corticotropin-releasing hormone through ERK1/2 pathway in murine melanoma cell line, B16F10. Exp. Dermatol. 2007, 16, 22–27. [Google Scholar] [CrossRef]
- Carlson, K.W.; Nawy, S.S.; Wei, E.T.; Sadee, W.; Filov, V.A.; Rezsova, V.V.; Slominski, A.; Quillan, J.M. Inhibition of mouse melanoma cell proliferation by corticotropin-releasing hormone and its analogs. Anticancer Res. 2001, 21, 1173–1179. [Google Scholar]
- Arnette, C.R.; Roth-Carter, Q.R.; Koetsier, J.L.; Broussard, J.A.; Burks, H.E.; Cheng, K.; Amadi, C.; Gerami, P.; Johnson, J.L.; Green, K.J. Keratinocyte cadherin desmoglein 1 controls melanocyte behavior through paracrine signaling. Pigment Cell Melanoma Res. 2020, 33, 305–317. [Google Scholar] [CrossRef]
- Liu, G.S.; Tsai, H.E.; Weng, W.T.; Liu, L.F.; Weng, C.H.; Chuang, M.R.; Lam, H.C.; Wu, C.S.; Tee, R.; Wen, Z.H.; et al. Systemic pro-opiomelanocortin expression induces melanogenic differentiation and inhibits tumor angiogenesis in established mouse melanoma. Hum. Gene Ther. 2011, 22, 325–335. [Google Scholar] [CrossRef]
- Wu, J.C.; Tsai, H.E.; Liu, G.S.; Wu, C.S.; Tai, M.H. Autophagic cell death participates in POMC-induced melanoma suppression. Cell Death Discov 2018, 4, 11. [Google Scholar] [CrossRef]
- Millington, G.W.M. Proopiomelanocortin (POMC): The cutaneous roles of its melanocortin products and receptors. Clin. Exp. Dermatol. 2006, 31, 407–412. [Google Scholar] [CrossRef]
- Pondeljak, N.; Lugović-Mihić, L. Stress-induced Interaction of Skin Immune Cells, Hormones, and Neurotransmitters. Clin. Ther. 2020, 42, 757–770. [Google Scholar] [CrossRef]
- Kadekaro, A.L.; Kavanagh, R.; Kanto, H.; Terzieva, S.; Hauser, J.; Kobayashi, N.; Schwemberger, S.; Cornelius, J.; Babcock, G.; Shertzer, H.G.; et al. alpha-Melanocortin and endothelin-1 activate antiapoptotic pathways and reduce DNA damage in human melanocytes. Cancer Res. 2005, 65, 4292–4299. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Malek, Z.A.; Knittel, J.; Kadekaro, A.L.; Swope, V.B.; Starner, R. The melanocortin 1 receptor and the UV response of human melanocytes--a shift in paradigm. Photochem. Photobiol. 2008, 84, 501–508. [Google Scholar] [CrossRef]
- Swope, V.B.; Abdel-Malek, Z.A. Significance of the Melanocortin 1 and Endothelin B Receptors in Melanocyte Homeostasis and Prevention of Sun-Induced Genotoxicity. Front. Genet. 2016, 7, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdomo, J.; Quintana, C.; González, I.; Hernández, I.; Rubio, S.; Loro, J.F.; Reiter, R.J.; Estévez, F.; Quintana, J. Melatonin Induces Melanogenesis in Human SK-MEL-1 Melanoma Cells Involving Glycogen Synthase Kinase-3 and Reactive Oxygen Species. Int. J. Mol. Sci. 2020, 21, 4970. [Google Scholar] [CrossRef]
- Liu, P.Y.; Johansson, O. Immunohistochemical evidence of alpha-, beta- and gamma 3-melanocyte stimulating hormone expression in cutaneous malignant melanoma of nodular type. J. Dermatol. Sci. 1995, 10, 203–212. [Google Scholar] [CrossRef]
- Nagahama, M.; Funasaka, Y.; Fernandez-Frez, M.L.; Ohashi, A.; Chakraborty, A.K.; Ueda, M.; Ichihashi, M. Immunoreactivity of alpha-melanocyte-stimulating hormone, adrenocorticotrophic hormone and beta-endorphin in cutaneous malignant melanoma and benign melanocytic naevi. Br. J. Dermatol. 1998, 138, 981–985. [Google Scholar] [CrossRef]
- Jiang, J.; Sharma, S.D.; Fink, J.L.; Hadley, M.E.; Hruby, V.J. Melanotropic peptide receptors: Membrane markers of human melanoma cells. Exp. Dermatol. 1996, 5, 325–333. [Google Scholar] [CrossRef]
- Palmer, J.S.; Duffy, D.L.; Box, N.F.; Aitken, J.F.; O’Gorman, L.E.; Green, A.C.; Hayward, N.K.; Martin, N.G.; Sturm, R.A. Melanocortin-1 receptor polymorphisms and risk of melanoma: Is the association explained solely by pigmentation phenotype? Am. J. Hum. Genet. 2000, 66, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Ichii-Jones, F.; Lear, J.T.; Heagerty, A.H.; Smith, A.G.; Hutchinson, P.E.; Osborne, J.; Bowers, B.; Jones, P.W.; Davies, E.; Ollier, W.E.; et al. Susceptibility to melanoma: Influence of skin type and polymorphism in the melanocyte stimulating hormone receptor gene. J. Investig. Dermatol. 1998, 111, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.E.; Rees, R.C.; MacNeil, S. A positive association between agonist-induced cyclic AMP production in vitro and metastatic potential in murine B16 melanoma and hamster fibrosarcoma. Clin. Exp. Metastasis 1990, 8, 461–474. [Google Scholar] [CrossRef]
- Eves, P.C.; MacNeil, S.; Haycock, J.W. alpha-Melanocyte stimulating hormone, inflammation and human melanoma. Peptides 2006, 27, 444–452. [Google Scholar] [CrossRef]
- Kameyama, K.; Vieira, W.D.; Tsukamoto, K.; Law, L.W.; Hearing, V.J. Differentiation and the tumorigenic and metastatic phenotype of murine melanoma cells. Int. J. Cancer 1990, 45, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Murata, J.; Ayukawa, K.; Ogasawara, M.; Fujii, H.; Saiki, I. Alpha-melanocyte-stimulating hormone blocks invasion of reconstituted basement membrane (Matrigel) by murine B16 melanoma cells. Invasion Metastasis 1997, 17, 82–93. [Google Scholar]
- Haycock, J.W.; Wagner, M.; Morandini, R.; Ghanem, G.; Rennie, I.G.; Mac Neil, S. Alpha-melanocyte-stimulating hormone inhibits NF-kappaB activation in human melanocytes and melanoma cells. J. Investig. Dermatol. 1999, 113, 560–566. [Google Scholar] [CrossRef]
- Katerinaki, E.; Haycock, J.W.; Lalla, R.; Carlson, K.E.; Yang, Y.; Hill, R.P.; Lorigan, P.C.; MacNeil, S. Sodium salicylate inhibits TNF-alpha-induced NF-kappaB activation, cell migration, invasion and ICAM-1 expression in human melanoma cells. Melanoma Res. 2006, 16, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, F.; Trentin, L.; Boyle, L.A.; Stamenkovic, I.; Byers, H.R.; Colvin, R.B.; Kurnick, J.T. Expression of cell adhesion molecules in human melanoma cell lines and their role in cytotoxicity mediated by tumor-infiltrating lymphocytes. Cancer 1992, 69, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Hedley, S.J.; Gawkrodger, D.J.; Weetman, A.P.; Morandini, R.; Boeynaems, J.M.; Ghanem, G.; Neil, S.M. alpha-Melanocyte stimulating hormone inhibits tumour necrosis factor-alpha stimulated intercellular adhesion molecule-1 expression in normal cutaneous human melanocytes and in melanoma cell lines. Br. J. Dermatol. 1998, 138, 536–543. [Google Scholar] [CrossRef]
- Morandini, R.; Boeynaems, J.M.; Hedley, S.J.; MacNeil, S.; Ghanem, G. Modulation of ICAM-1 expression by alpha-MSH in human melanoma cells and melanocytes. J. Cell. Physiol. 1998, 175, 276–282. [Google Scholar] [CrossRef]
- Natali, P.; Nicotra, M.R.; Cavaliere, R.; Bigotti, A.; Romano, G.; Temponi, M.; Ferrone, S. Differential expression of intercellular adhesion molecule 1 in primary and metastatic melanoma lesions. Cancer Res. 1990, 50, 1271–1278. [Google Scholar] [PubMed]
- Johnson, J.P. The role of ICAM-1 in tumor development. Chem. Immunol. 1991, 50, 143–163. [Google Scholar] [PubMed]
- Eves, P.; Haycock, J.; Layton, C.; Wagner, M.; Kemp, H.; Szabo, M.; Morandini, R.; Ghanem, G.; García-Borrón, J.C.; Jiménez-Cervantes, C.; et al. Anti-inflammatory and anti-invasive effects of alpha-melanocyte-stimulating hormone in human melanoma cells. Br. J. Cancer 2003, 89, 2004–2015. [Google Scholar] [CrossRef]
- Zhu, N.; Eves, P.C.; Katerinaki, E.; Szabo, M.; Morandini, R.; Ghanem, G.; Lorigan, P.; MacNeil, S.; Haycock, J.W. Melanoma cell attachment, invasion, and integrin expression is upregulated by tumor necrosis factor alpha and suppressed by alpha melanocyte stimulating hormone. J. Investig. Dermatol. 2002, 119, 1165–1171. [Google Scholar] [CrossRef]
- Canton, I.; Eves, P.C.; Szabo, M.; Vidal-Vanaclocha, F.; Sisley, K.; Rennie, I.G.; Haycock, J.W.; MacNeil, S. Tumor necrosis factor alpha increases and alpha-melanocyte-stimulating hormone reduces uveal melanoma invasion through fibronectin. J. Investig. Dermatol. 2003, 121, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Farzanefar, S.; Etemadi, R.; Shirkhoda, M.; Mahmoodzadeh, H.; Erfani, M.; Fallahi, B.; Abbasi, M.; Ayati, N.; Hassanzadeh-Rad, A.; Eftekhari, M.; et al. The Value of Technetium-99m Labeled Alpha-Melanocyte-Stimulating Hormone ((99m)Tc-α-MSH) in Diagnosis of Primary and Metastatic Lesions of Malignant Melanoma. Asia Ocean J. Nucl. Med. Biol 2018, 6, 155–160. [Google Scholar] [CrossRef]
- Gao, F.; Sihver, W.; Bergmann, R.; Belter, B.; Bolzati, C.; Salvarese, N.; Steinbach, J.; Pietzsch, J.; Pietzsch, H.J. Synthesis, Characterization, and Initial Biological Evaluation of [(99m) Tc]Tc-Tricarbonyl-labeled DPA-α-MSH Peptide Derivatives for Potential Melanoma Imaging. ChemMedChem 2018, 13, 1146–1158. [Google Scholar] [CrossRef]
- Xu, J.; Yang, J.; Gonzalez, R.; Fisher, D.R.; Miao, Y. Melanoma-Targeting Property of Y-90-Labeled Lactam-Cyclized α-Melanocyte-Stimulating Hormone Peptide. Cancer Biother. Radiopharm. 2019, 34, 597–603. [Google Scholar] [CrossRef]
- Palangka, C.R.A.P.; Hanaoka, H.; Yamaguchi, A.; Murakami, T.; Tsushima, Y. Al18F-labeled alpha-melanocyte-stimulating hormone (α-MSH) peptide derivative for the early detection of melanoma. Ann. Nucl. Med. 2019, 33, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, Z.; Lin, K.S.; Lau, J.; Zeisler, J.; Colpo, N.; Perrin, D.M.; Bénard, F. Melanoma Imaging Using (18)F-Labeled α-Melanocyte-Stimulating Hormone Derivatives with Positron Emission Tomography. Mol. Pharm. 2018, 15, 2116–2122. [Google Scholar] [CrossRef]
- Gao, F.; Sihver, W.; Bergmann, R.; Walther, M.; Stephan, H.; Belter, B.; Neuber, C.; Haase-Kohn, C.; Bolzati, C.; Pietzsch, J.; et al. Radiochemical and radiopharmacological characterization of a 64Cu-labeled α-MSH analog conjugated with different chelators. J. Label. Compd. Radiopharm. 2019, 62, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Kato, T.; Washiyama, K.; Ihara, M.; Mizutani, A.; Nishi, K.; Flores, L.G., 2nd; Nishii, R.; Kawai, K. The pharmacological properties of 3-arm or 4-arm DOTA constructs for conjugation to α-melanocyte-stimulating hormone analogues for melanoma imaging. PLoS ONE 2019, 14, e0213397. [Google Scholar] [CrossRef]
- Yang, J.; Xu, J.; Cheuy, L.; Gonzalez, R.; Fisher, D.R.; Miao, Y. Evaluation of a Novel Pb-203-Labeled Lactam-Cyclized Alpha-Melanocyte-Stimulating Hormone Peptide for Melanoma Targeting. Mol. Pharm. 2019, 16, 1694–1702. [Google Scholar] [CrossRef]
- Zhao, Y.; Pang, B.; Detering, L.; Luehmann, H.; Yang, M.; Black, K.; Sultan, D.; Xia, Y.; Liu, Y. Melanocortin 1 Receptor Targeted Imaging of Melanoma With Gold Nanocages and Positron Emission Tomography. Mol. Imaging 2018, 17, 1536012118775827. [Google Scholar] [CrossRef]
- Chakraborty, A.; Slominski, A.; Ermak, G.; Hwang, J.; Pawelek, J. Ultraviolet B and Melanocyte-Stimulating Hormone (MSH) Stimulate mRNA Production for ∝MSH Receptors and Proopiomelanocortin-Derived Peptides in Mouse Melanoma Cells and Transformed Keratinocytes. J. Investig. Dermatol. 1995, 105, 655–659. [Google Scholar] [CrossRef]
- Hedley, S.J.; Murray, A.; Sisley, K.; Ghanem, G.; Morandini, R.; Gawkrodger, D.J.; Mac Neil, S. Alpha-melanocyte stimulating hormone can reduce T-cell interaction with melanoma cells in vitro. Melanoma Res. 2000, 10, 323–330. [Google Scholar] [CrossRef]
- Haycock, J.W.; Wagner, M.; Morandini, R.; Ghanem, G.; Rennie, I.G.; MacNeil, S. alpha-MSH immunomodulation acts via rel/NF-kappa B in cutaneous and ocular melanocytes and in melanoma cells. Ann. N. Y. Acad. Sci. 1999, 885, 396–399. [Google Scholar] [CrossRef]
- Ghanem, G.; Verstegen, J.; Libert, A.; Arnould, R.; Lejeune, F. Alpha-melanocyte-stimulating hormone immunoreactivity in human melanoma metastases extracts. Pigment Cell Res. 1989, 2, 519–523. [Google Scholar] [CrossRef]
- Lunec, J.; Pieron, C.; Sherbet, G.V.; Thody, A.J. Alpha-melanocyte-stimulating hormone immunoreactivity in melanoma cells. Pathobiology 1990, 58, 193–197. [Google Scholar] [CrossRef]
- Loir, B.; Bouchard, B.; Morandini, R.; Del Marmol, V.; Deraemaecker, R.; Garcia-Borron, J.C.; Ghanem, G. Immunoreactive alpha-melanotropin as an autocrine effector in human melanoma cells. Eur. J. Biochem. 1997, 244, 923–930. [Google Scholar] [CrossRef]
- Gáspár, E.; Nguyen-Thi, K.T.; Hardenbicker, C.; Tiede, S.; Plate, C.; Bodó, E.; Knuever, J.; Funk, W.; Bíró, T.; Paus, R. Thyrotropin-Releasing Hormone Selectively Stimulates Human Hair Follicle Pigmentation. J. Investig. Dermatol. 2011, 131, 2368–2377. [Google Scholar] [CrossRef] [Green Version]
- Schioth, H.B.; Prusis, P.; Muceniece, R.; Mutulis, F.; Mutule, I.; Wikberg, J.E. Thyrotropin releasing hormone (TRH) selectively binds and activates the melanocortin 1 receptor. Peptides 1999, 20, 395–400. [Google Scholar] [CrossRef]
- Ellerhorst, J.A.; Naderi, A.A.; Johnson, M.K.; Pelletier, P.; Prieto, V.G.; Diwan, A.H.; Johnson, M.M.; Gunn, D.C.; Yekell, S.; Grimm, E.A. Expression of thyrotropin-releasing hormone by human melanoma and nevi. Clin. Cancer Res. 2004, 10, 5531–5536. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, E.F.; Engel, W.K.; Askanas, V. Cells of neural crest origin as possible models to investigate thyrotropin releasing hormone action in the central nervous system. Neuropharmacology 1985, 24, 1109–1112. [Google Scholar] [CrossRef]
- Wilber, J.F.; Spinella, P. Identification of immunoreactive thyrotropin-releasing hormone in human neoplasia. J. Clin. Endocrinol. Metab. 1984, 59, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Lum, S.S.; Fletcher, W.S.; O’Dorisio, M.S.; Nance, R.W.; Pommier, R.F.; Caprara, M. Distribution and functional significance of somatostatin receptors in malignant melanoma. World J. Surg. 2001, 25, 407–412. [Google Scholar] [CrossRef]
- Martinez-Alonso, M.; Llecha, N.; Mayorga, M.E.; Sorolla, A.; Dolcet, X.; Sanmartin, V.; Abal, L.; Casanova, J.M.; Baradad, M.; Yeramian, A.; et al. Expression of somatostatin receptors in human melanoma cell lines: Effect of two different somatostatin analogues, octreotide and SOM230, on cell proliferation. J. Int. Med. Res. 2009, 37, 1813–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, I.; De, M.; Dey, G.; Bharti, R.; Chattopadhyay, S.; Ali, N.; Chakrabarti, P.; Reis, R.L.; Kundu, S.C.; Mandal, M. A peptide-modified solid lipid nanoparticle formulation of paclitaxel modulates immunity and outperforms dacarbazine in a murine melanoma model. Biomater. Sci. 2019, 7, 1161–1178. [Google Scholar] [CrossRef]
- Dummer, R.; Michielin, O.; Nägeli, M.C.; Goldinger, S.M.; Campigotto, F.; Kriemler-Krahn, U.; Schmid, H.; Pedroncelli, A.; Micaletto, S.; Schadendorf, D. Phase I, open-label study of pasireotide in patients with BRAF-wild type and NRAS-wild type, unresectable and/or metastatic melanoma. ESMO Open 2018, 3, e000388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harda, K.; Szabo, Z.; Szabo, E.; Olah, G.; Fodor, K.; Szasz, C.; Mehes, G.; Schally, A.V.; Halmos, G. Somatostatin Receptors as Molecular Targets in Human Uveal Melanoma. Molecules 2018, 23, 1535. [Google Scholar] [CrossRef] [Green Version]
- Ardjomand, N.; Ardjomand, N.; Schaffler, G.; Radner, H.; El-Shabrawi, Y. Expression of somatostatin receptors in uveal melanomas. Investig. Ophthalmol. Vis. Sci. 2003, 44, 980–987. [Google Scholar] [CrossRef] [Green Version]
- Kouch-el Filali, M.; Kilic, E.; Melis, M.; de Klein, A.; de Jong, M.; Luyten, G.P. Expression of the SST receptor 2 in uveal melanoma is not a prognostic marker. Graefes Arch. Clin. Exp. Ophthalmol. 2008, 246, 1585–1592. [Google Scholar] [CrossRef]
- Bankir, L.; Bichet, D.G.; Morgenthaler, N.G. Vasopressin: Physiology, assessment and osmosensation. J. Intern. Med. 2017, 282, 284–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, N.; Martins, D.; Santos, A.J.; Prata, D.; Veríssimo, M. How do hypothalamic nonapeptides shape youth’s sociality? A systematic review on oxytocin, vasopressin and human socio-emotional development. Neurosci. Biobehav. Rev. 2018, 90, 309–331. [Google Scholar] [CrossRef]
- Aroni, K.; Charitopoulos, K.N.; Lazaris, A.C.; Davaris, P. Lack of vasopressin expression in malignant melanomas. Melanoma Res. 2000, 10, 535. [Google Scholar] [CrossRef]
- Ripoll, G.V.; Farina, H.G.; Yoshiji, H.; Gomez, D.E.; Alonso, D.F. Desmopressin reduces melanoma lung metastasis in transgenic mice overexpressing tissue inhibitor of metalloproteinases-1. Vivo 2006, 20, 881–885. [Google Scholar]
- Li, C.; Kim, K. Neuropeptides. Wormbook Online Rev. C Elegans Biol. 2008, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Solomon, I.; Voiculescu, V.M.; Caruntu, C.; Lupu, M.; Popa, A.; Ilie, M.A.; Albulescu, R.; Caruntu, A.; Tanase, C.; Constantin, C.; et al. Neuroendocrine Factors and Head and Neck Squamous Cell Carcinoma: An Affair to Remember. Dis. Markers 2018, 2018, 9787831. [Google Scholar] [CrossRef] [Green Version]
- Khare, V.K.; Albino, A.P.; Reed, J.A. The neuropeptide/mast cell secretagogue substance P is expressed in cutaneous melanocytic lesions. J. Cutan. Pathol. 1998, 25, 2–10. [Google Scholar] [CrossRef]
- Borrego, J.F.; Huelsmeyer, M.K.; Pinkerton, M.E.; Muszynski, J.L.; Miller, S.A.; Kurzman, I.D.; Vail, D.M. Neurokinin-1 receptor expression and antagonism by the NK-1R antagonist maropitant in canine melanoma cell lines and primary tumour tissues. Vet. Comp. Oncol. 2016, 14, 210–224. [Google Scholar] [CrossRef]
- Muñoz, M.; Rosso, M.; Robles-Frias, M.J.; Salinas-Martín, M.V.; Rosso, R.; González-Ortega, A.; Coveñas, R. The NK-1 receptor is expressed in human melanoma and is involved in the antitumor action of the NK-1 receptor antagonist aprepitant on melanoma cell lines. Lab. Investig. 2010, 90, 1259–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, X.; Li, J.; Hu, H.; Miao, X.; Song, X.; Yang, W.; Zeng, Q.; Mou, L.; Wang, R. Human hemokinin-1 promotes migration of melanoma cells and increases MMP-2 and MT1-MMP expression by activating tumor cell NK1 receptors. Peptides 2016, 83, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Rosso, M. The NK-1 receptor antagonist aprepitant as a broad spectrum antitumor drug. Investig. New Drugs 2010, 28, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Pérez, A.; Rosso, M.; Zamarriego, C.; Rosso, R. Antitumoral action of the neurokinin-1 receptor antagonist L-733 060 on human melanoma cell lines. Melanoma Res. 2004, 14, 183–188. [Google Scholar] [CrossRef] [PubMed]
- González-Ortega, A.; Sánchez-Vaderrábanos, E.; Ramiro-Fuentes, S.; Salinas-Martín, M.V.; Carranza, A.; Coveñas, R.; Muñoz, M. Uveal melanoma expresses NK-1 receptors and cyclosporin A induces apoptosis in human melanoma cell lines overexpressing the NK-1 receptor. Peptides 2014, 55, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Bernabeu-Wittel, J.; Coveñas, R. NK-1 as a melanoma target. Expert Opin. Ther. Targets 2011, 15, 889–897. [Google Scholar] [CrossRef]
- Scheau, C.; Badarau, I.A.; Costache, R.; Caruntu, C.; Mihai, G.L.; Didilescu, A.C.; Constantin, C.; Neagu, M. The Role of Matrix Metalloproteinases in the Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma. Anal. Cell. Pathol. 2019, 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Matalińska, J.; Lipiński, P.F.J.; Kosson, P.; Kosińska, K.; Misicka, A. In Vivo, In Vitro and In Silico Studies of the Hybrid Compound AA3266, an Opioid Agonist/NK1R Antagonist with Selective Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 7738. [Google Scholar] [CrossRef]
- Janecka, A.; Poels, J.; Fichna, J.; Studzian, K.; Vanden Broeck, J. Comparison of antagonist activity of spantide family at human neurokinin receptors measured by aequorin luminescence-based functional calcium assay. Regul. Pept. 2005, 131, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Ping, F.; Shang, J.; Zhou, J.; Song, J.; Zhang, L. Activation of neurokinin-1 receptor by substance P inhibits melanogenesis in B16-F10 melanoma cells. Int. J. Biochem. Cell Biol. 2012, 44, 2342–2348. [Google Scholar] [CrossRef]
- Korcum, A.F.; Sanlioglu, S.; Aksu, G.; Tuncel, N.; Erin, N. Radiotherapy-induced decreases in substance P levels may potentiate melanoma growth. Mol. Med. Rep 2009, 2, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Zabel, M.; Dietel, M.; Gębarowska, E.; Michael, R. Effect of Follicular Cells on Calcitonin Gene Expression in Thyroid Parafollicular Cells in Cell Culture. Histochem. J. 1999, 31, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, M.; Luo, Y.; Makino, T.; Matsui, C.; Morohashi, M. Calcitonin gene-related peptide upregulates melanogenesis and enhances melanocyte dendricity via induction of keratinocyte-derived melanotrophic factors. J. Investig. Dermatol. Symp. Proc. 1999, 4, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Feng, J.Y.; Wang, Q.; Shang, J. Calcitonin gene-related peptide cooperates with substance P to inhibit melanogenesis and induces apoptosis of B16F10 cells. Cytokine 2015, 74, 137–144. [Google Scholar] [CrossRef]
- Golias, C.; Charalabopoulos, A.; Stagikas, D.; Charalabopoulos, K.; Batistatou, A. The kinin system--bradykinin: Biological effects and clinical implications. Multiple role of the kinin system--bradykinin. Hippokratia 2007, 11, 124–128. [Google Scholar] [PubMed]
- Liu, H.-T.; Akita, T.; Shimizu, T.; Sabirov, R.Z.; Okada, Y. Bradykinin-induced astrocyte-neuron signalling: Glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. J. Physiol. 2009, 587, 2197–2209. [Google Scholar] [CrossRef]
- Ikeda, Y.; Hayashi, I.; Kamoshita, E.; Yamazaki, A.; Endo, H.; Ishihara, K.; Yamashina, S.; Tsutsumi, Y.; Matsubara, H.; Majima, M. Host stromal bradykinin B2 receptor signaling facilitates tumor-associated angiogenesis and tumor growth. Cancer Res. 2004, 64, 5178–5185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunn, P.A., Jr.; Chan, D.; Dienhart, D.G.; Tolley, R.; Tagawa, M.; Jewett, P.B. Neuropeptide signal transduction in lung cancer: Clinical implications of bradykinin sensitivity and overall heterogeneity. Cancer Res. 1992, 52, 24–31. [Google Scholar]
- Fujita, M.; Andoh, T.; Ohashi, K.; Akira, A.; Saiki, I.; Kuraishi, Y. Roles of kinin B1 and B2 receptors in skin cancer pain produced by orthotopic melanoma inoculation in mice. Eur. J. Pain 2010, 14, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Raidoo, D.M.; Sawant, S.; Mahabeer, R.; Bhoola, K.D. Kinin receptors are expressed in human astrocytic tumour cells. Immunopharmacology 1999, 43, 255–263. [Google Scholar] [CrossRef]
- Wu, J.; Akaike, T.; Hayashida, K.; Miyamoto, Y.; Nakagawa, T.; Miyakawa, K.; Muller-Esterl, W.; Maeda, H. Identification of bradykinin receptors in clinical cancer specimens and murine tumor tissues. Int. J. Cancer 2002, 98, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Andoh, T.; Akira, A.; Saiki, I.; Kuraishi, Y. Bradykinin increases the secretion and expression of endothelin-1 through kinin B2 receptors in melanoma cells. Peptides 2010, 31, 238–241. [Google Scholar] [CrossRef]
- Andoh, T.; Shinohara, A.; Kuraishi, Y. Inhibitory effect of fentanyl citrate on the release of endothlin-1 induced by bradykinin in melanoma cells. Pharmacol. Rep. 2017, 69, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Maria, A.G.; Dillenburg-Pilla, P.; Reis, R.I.; Floriano, E.M.; Tefe-Silva, C.; Ramos, S.G.; Pesquero, J.B.; Nahmias, C.; Costa-Neto, C.M. Host kinin B1 receptor plays a protective role against melanoma progression. Sci. Rep. 2016, 6, 22078. [Google Scholar] [CrossRef] [PubMed]
- Dillenburg-Pilla, P.; Maria, A.G.; Reis, R.I.; Floriano, E.M.; Pereira, C.D.; De Lucca, F.L.; Ramos, S.G.; Pesquero, J.B.; Jasiulionis, M.G.; Costa-Neto, C.M. Activation of the kinin B1 receptor attenuates melanoma tumor growth and metastasis. PLoS ONE 2013, 8, e64453. [Google Scholar] [CrossRef] [Green Version]
- Maria, A.G.; Dillemburg-Pilla, P.; Durand, M.T.; Floriano, E.M.; Manfiolli, A.O.; Ramos, S.G.; Pesquero, J.B.; Nahmias, C.; Costa-Neto, C.M. Activation of the Kinin B1 Receptor by Its Agonist Reduces Melanoma Metastasis by Playing a Dual Effect on Tumor Cells and Host Immune Response. Front. Pharmacol. 2019, 10, 1106. [Google Scholar] [CrossRef] [PubMed]
- Beck, B. Neuropeptide Y in normal eating and in genetic and dietary-induced obesity. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2006, 361, 1159–1185. [Google Scholar] [CrossRef] [Green Version]
- Gilaberte, Y.; Roca, M.J.; Garcia-Prats, M.D.; Coscojuela, C.; Arbues, M.D.; Vera-Alvarez, J.J. Neuropeptide Y expression in cutaneous melanoma. J. Am. Acad. Dermatol. 2012, 66, e201–e208. [Google Scholar] [CrossRef]
- Pérez Tato, B.; Juarranz, Á.; Nájera, L.; Mihm, M.C.; Fernández, P.; Gilaberte, Y.; González, S. Neuropeptide Y expression in primary cutaneous melanoma. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 443–449. [Google Scholar] [CrossRef]
- Alasvand, M.; Rashidi, B.; Javanmard, S.H.; Akhavan, M.M.; Khazaei, M. Effect of Blocking of Neuropeptide Y Y2 Receptor on Tumor Angiogenesis and Progression in Normal and Diet-Induced Obese C57BL/6 Mice. Glob. J. Health Sci. 2015, 7, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louridas, M.; Letourneau, S.; Lautatzis, M.-E.; Vrontakis, M. Galanin is highly expressed in bone marrow mesenchymal stem cells and facilitates migration of cells both in vitro and in vivo. Biochem. Biophys. Res. Commun. 2009, 390, 867–871. [Google Scholar] [CrossRef]
- Santha, P.; Pierau, F.K.; Jancso, G. Galanin mediated inhibitory nervous modulation of cutaneous vascular reactions. Acta Physiol. Hung. 1999, 86, 279–285. [Google Scholar]
- Holmberg, K.; Kuteeva, E.; Brumovsky, P.; Kahl, U.; Karlström, H.; Lucas, G.A.; Rodriguez, J.; Westerblad, H.; Hilke, S.; Theodorsson, E.; et al. Generation and phenotypic characterization of a galanin overexpressing mouse. Neuroscience 2005, 133, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.W.; Lang, R.; Jakab, M.; Kofler, B. Galanin family of peptides in skin function. Cell. Mol. Life Sci. 2008, 65, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Zhang, X.; Zhang, Q.; Dagerlind, A.; Nilsson, S.; Wiesenfeld-Hallin, Z.; Hökfelt, T. Central and peripheral expression of galanin in response to inflammation. Neuroscience 1995, 68, 563–576. [Google Scholar] [CrossRef]
- Berger, A.; Santic, R.; Hauser-Kronberger, C.; Schilling, F.H.; Kogner, P.; Ratschek, M.; Gamper, A.; Jones, N.; Sperl, W.; Kofler, B. Galanin and galanin receptors in human cancers. Neuropeptides 2005, 39, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Lang, R.; Moritz, K.; Santic, R.; Hermann, A.; Sperl, W.; Kofler, B. Galanin receptor subtype GalR2 mediates apoptosis in SH-SY5Y neuroblastoma cells. Endocrinology 2004, 145, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Kofler, B.; Berger, A.; Santic, R.; Moritz, K.; Almer, D.; Tuechler, C.; Lang, R.; Emberger, M.; Klausegger, A.; Sperl, W.; et al. Expression of neuropeptide galanin and galanin receptors in human skin. J. Investig. Dermatol. 2004, 122, 1050–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henson, B.S.; Neubig, R.R.; Jang, I.; Ogawa, T.; Zhang, Z.; Carey, T.E.; D’Silva, N.J. Galanin receptor 1 has anti-proliferative effects in oral squamous cell carcinoma. J. Biol. Chem. 2005, 280, 22564–22571. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, T.; Iwashita, T.; Kommareddi, P.; Nair, T.; Misawa, K.; Misawa, Y.; Ueda, Y.; Tono, T.; Carey, T.E. Galanin and galanin receptor type 1 suppress proliferation in squamous carcinoma cells: Activation of the extracellular signal regulated kinase pathway and induction of cyclin-dependent kinase inhibitors. Oncogene 2007, 26, 5762–5771. [Google Scholar] [CrossRef] [Green Version]
- Gilaberte, Y.; Vera, J.; Coscojuela, C.; Roca, M.J.; Parrado, C.; Gonzalez, S. Expression of galanin in melanocytic tumors. Actas Dermo Sifiliogr. 2007, 98, 24–34. [Google Scholar] [CrossRef]
- Charitopoulos, K.N.; Lazaris, A.C.; Aroni, K.; Kavantzas, N.; Nikolakopoulou, E.; Davaris, P. Immunodetection of gastrin-releasing peptide in malignant melanoma cells. Melanoma Res. 2000, 10, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Marrone, B.F.; Meurer, L.; Moretto, A.; Kleina, W.; Schwartsmann, G. Expression of gastrin-releasing peptide receptor in patients with cutaneous malignant melanoma. Clin. Exp. Dermatol. 2013, 38, 707–712. [Google Scholar] [CrossRef]
- Pansky, A.; Peng, F.; Eberhard, M.; Baselgia, L.; Siegrist, W.; Baumann, J.B.; Eberle, A.N.; Beglinger, C.; Hildebrand, P. Identification of functional GRP-preferring bombesin receptors on human melanoma cells. Eur. J. Clin. Investig. 1997, 27, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, X.; Wang, R.; Liu, S.; Wang, Y.; Jing, L.; Louis, M.D.J.; Cao, R. Comparison of fusion protein and DC vaccine in inhibition of mouse B16F10 melanoma tumor. Biomed. Pharmacother. 2018, 97, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Cullen, J.M.; Cascella, M. Physiology, Enkephalin. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Takahashi, A. Subchapter 7A-Enkephalin. In Handbook of Hormones; Takei, Y., Ando, H., Tsutsui, K., Eds.; Academic Press: San Diego, CA, USA, 2016; p. 55-e57A-52. [Google Scholar] [CrossRef]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [Green Version]
- Azzam, A.A.H.; McDonald, J.; Lambert, D.G. Hot topics in opioid pharmacology: Mixed and biased opioids. Br. J. Anaesth. 2019, 122, e136–e145. [Google Scholar] [CrossRef]
- Bigliardi, P.L.; Tobin, D.J.; Gaveriaux-Ruff, C.; Bigliardi-Qi, M. Opioids and the skin–where do we stand? Exp. Dermatol. 2009, 18, 424–430. [Google Scholar] [CrossRef]
- Bisignani, G.J.; McLaughlin, P.J.; Ordille, S.D.; Beltz, M.S.; Jarowenko, M.V.; Zagon, I.S. Human renal cell cancer proliferation in tissue culture is tonically inhibited by opioid growth factor. J. Urol. 1999, 162, 2186–2191. [Google Scholar] [CrossRef]
- Slominski, A.T.; Zmijewski, M.A.; Zbytek, B.; Brozyna, A.A.; Granese, J.; Pisarchik, A.; Szczesniewski, A.; Tobin, D.J. Regulated proenkephalin expression in human skin and cultured skin cells. J. Investig. Dermatol. 2011, 131, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Henry, M.S.; Gendron, L.; Tremblay, M.-E.; Drolet, G. Enkephalins: Endogenous Analgesics with an Emerging Role in Stress Resilience. Neural Plast. 2017, 2017, 1546125. [Google Scholar] [CrossRef] [PubMed]
- Toubia, T.; Khalife, T. The Endogenous Opioid System: Role and Dysfunction Caused by Opioid Therapy. Clin. Obstet. Gynecol. 2019, 62, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; Smith, J.P.; McLaughlin, P.J. Human pancreatic cancer cell proliferation in tissue culture is tonically inhibited by opioid growth factor. Int. J. Oncol. 1999, 14, 577–584. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, P.J.; Levin, R.J.; Zagon, I.S. Regulation of human head and neck squamous cell carcinoma growth in tissue culture by opioid growth factor. Int. J. Oncol. 1999, 14, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Donahue, R.N.; McLaughlin, P.J.; Zagon, I.S. Cell proliferation of human ovarian cancer is regulated by the opioid growth factor-opioid growth factor receptor axis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1716–R1725. [Google Scholar] [CrossRef] [Green Version]
- Zagon, I.S.; Donahue, R.N.; McLaughlin, P.J. Opioid growth factor-opioid growth factor receptor axis is a physiological determinant of cell proliferation in diverse human cancers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1154–R1161. [Google Scholar] [CrossRef] [Green Version]
- Murgo, A.J. Inhibition of B16-BL6 melanoma growth in mice by methionine-enkephalin. J. Natl. Cancer Inst. 1985, 75, 341–344. [Google Scholar] [PubMed]
- Wang, D.M.; Jiao, X.; Plotnikoff, N.P.; Griffin, N.; Qi, R.Q.; Gao, X.H.; Shan, F.P. Killing effect of methionine enkephalin on melanoma in vivo and in vitro. Oncol. Rep. 2017, 38, 2132–2140. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.-M.; Wang, G.-C.; Yang, J.; Plotnikoff, N.P.; Griffin, N.; Han, Y.-M.; Qi, R.-Q.; Gao, X.-H.; Shan, F.-P. Inhibition of the growth of human melanoma cells by methionine enkephalin. Mol. Med. Rep. 2016, 14, 5521–5527. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.W.; Wang, S.T.; Chang, S.H.; Chuang, K.C.; Wang, H.Y.; Kao, J.K.; Liang, S.M.; Wu, C.Y.; Kao, S.H.; Chen, Y.J.; et al. Imiquimod Exerts Antitumor Effects by Inducing Immunogenic Cell Death and Is Enhanced by the Glycolytic Inhibitor 2-Deoxyglucose. J. Investig. Dermatol. 2020, 140, 1771–1783.e6. [Google Scholar] [CrossRef]
- Zagon, I.S.; Donahue, R.N.; Rogosnitzky, M.; McLaughlin, P.J. Imiquimod upregulates the opioid growth factor receptor to inhibit cell proliferation independent of immune function. Exp. Biol. Med. 2008, 233, 968–979. [Google Scholar] [CrossRef]
- O’Hern, K.; Chambers, M.; Ryan, C.; Chapman, M.S. In lieu of penectomy: Complete resolution of penile melanoma in situ with topical imiquimod and tretinoin. Int. J. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nahm, W.J.; Gwillim, E.C.; Badiavas, E.V.; Nichols, A.J.; Kirsner, R.S.; Boggeln, L.H.; Shen, J.T. Treating Melanoma in Situ During a Pandemic with Telemedicine and a Combination of Imiquimod, 5-Fluorouracil, and Tretinoin. Dermatol. Ther. 2021, 11, 307–314. [Google Scholar] [CrossRef]
- Iznardo, H.; Garcia-Melendo, C.; Yélamos, O. Lentigo Maligna: Clinical Presentation and Appropriate Management. Clin. Cosmet. Investig. Dermatol. 2020, 13, 837–855. [Google Scholar] [CrossRef]
- Lobo, Y.; Templeman, R. Conservative treatment of lentigo maligna with topical imiquimod 5% cream: A case report. Dermatol. Online J. 2020, 26. [Google Scholar]
- Scarfì, F.; Patrizi, A.; Veronesi, G.; Lambertini, M.; Tartari, F.; Mussi, M.; Melotti, B.; Dika, E. The role of topical imiquimod in melanoma cutaneous metastases: A critical review of the literature. Dermatol. Ther. 2020, 33, e14165. [Google Scholar] [CrossRef]
- Machelska, H.; Celik, M. Opioid Receptors in Immune and Glial Cells-Implications for Pain Control. Front. Immunol. 2020, 11, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, I.; Sierra, S.; Lueptow, L.; Gupta, A.; Gouty, S.; Margolis, E.B.; Cox, B.M.; Devi, L.A. Biased signaling by endogenous opioid peptides. Proc. Natl. Acad. Sci. USA 2020, 117, 11820–11828. [Google Scholar] [CrossRef]
- Pilozzi, A.; Carro, C.; Huang, X. Roles of β-Endorphin in Stress, Behavior, Neuroinflammation, and Brain Energy Metabolism. Int. J. Mol. Sci. 2020, 22, 338. [Google Scholar] [CrossRef]
- Okano, T.; Sato, K.; Shirai, R.; Seki, T.; Shibata, K.; Yamashita, T.; Koide, A.; Tezuka, H.; Mori, Y.; Hirano, T.; et al. β-Endorphin Mediates the Development and Instability of Atherosclerotic Plaques. Int. J. Endocrinol. 2020, 2020, 4139093. [Google Scholar] [CrossRef]
- Qiu, J.; Jiang, Y.F.; Li, F.; Tong, Q.H.; Rong, H.; Cheng, R. Effect of combined music and touch intervention on pain response and β-endorphin and cortisol concentrations in late preterm infants. Bmc Pediatr. 2017, 17, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corder, G.; Castro, D.C.; Bruchas, M.R.; Scherrer, G. Endogenous and Exogenous Opioids in Pain. Annu. Rev. Neurosci. 2018, 41, 453–473. [Google Scholar] [CrossRef]
- Mousa, S.A.; Zhang, Q.; Sitte, N.; Ji, R.-R.; Stein, C. β-Endorphin-containing memory-cells and μ-opioid receptors undergo transport to peripheral inflamed tissue. J. Neuroimmunol. 2001, 115, 71–78. [Google Scholar] [CrossRef]
- Luan, Y.H.; Wang, D.; Yu, Q.; Chai, X.Q. Action of β-endorphin and nonsteroidal anti-inflammatory drugs, and the possible effects of nonsteroidal anti-inflammatory drugs on β-endorphin. J. Clin. Anesth. 2017, 37, 123–128. [Google Scholar] [CrossRef]
- Sarkar, D.K.; Murugan, S.; Zhang, C.; Boyadjieva, N. Regulation of cancer progression by β-endorphin neuron. Cancer Res. 2012, 72, 836–840. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, D.K.; Boyadjieva, N.I.; Chen, C.P.; Ortigüela, M.; Reuhl, K.; Clement, E.M.; Kuhn, P.; Marano, J. Cyclic adenosine monophosphate differentiated beta-endorphin neurons promote immune function and prevent prostate cancer growth. Proc. Natl. Acad. Sci. USA 2008, 105, 9105–9110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, J.E.; Schwertfeger, K.L. Proinflammatory cytokines in breast cancer: Mechanisms of action and potential targets for therapeutics. Curr. Drug Targets 2010, 11, 1133–1146. [Google Scholar] [CrossRef]
- Argueta, D.A.; Aich, A.; Lei, J.; Kiven, S.; Nguyen, A.; Wang, Y.; Gu, J.; Zhao, W.; Gupta, K. β-endorphin at the intersection of pain and cancer progression: Preclinical evidence. Neurosci. Lett. 2021, 744, 135601. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, S.; Hardt, K.; Schadendorf, D.; Henschler, R.; Boehncke, W.H.; Duthey, B. Endogenous μ-opioid peptides modulate immune response towards malignant melanoma. Exp. Dermatol. 2011, 20, 24–28. [Google Scholar] [CrossRef]
- Slominski, A. Identification of beta-endorphin, alpha-MSH and ACTH peptides in cultured human melanocytes, melanoma and squamous cell carcinoma cells by RP-HPLC. Exp. Dermatol. 1998, 7, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Fell, G.L.; Robinson, K.C.; Mao, J.; Woolf, C.J.; Fisher, D.E. Skin β-endorphin mediates addiction to UV light. Cell 2014, 157, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Delgado, M.; Ganea, D. Vasoactive intestinal peptide: A neuropeptide with pleiotropic immune functions. Amino Acids 2013, 45, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.H.; Yao, C.; Oh, J.H.; Park, C.H.; Tian, Y.D.; Han, M.; Kim, J.E.; Chung, J.H.; Jin, Z.H.; Lee, D.H. Vasoactive intestinal peptide stimulates melanogenesis in B16F10 mouse melanoma cells via CREB/MITF/tyrosinase signaling. Biochem. Biophys. Res. Commun. 2016, 477, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Antohe, M.; Nedelcu, R.I.; Nichita, L.; Popp, C.G.; Cioplea, M.; Brinzea, A.; Hodorogea, A.; Calinescu, A.; Balaban, M.; Ion, D.A.; et al. Tumor infiltrating lymphocytes: The regulator of melanoma evolution. Oncol. Lett. 2019, 17, 4155–4161. [Google Scholar] [CrossRef]
- Căruntu, C.; Boda, D.; Musat, S.; Căruntu, A.; Mandache, E. Stress-induced mast cell activation in glabrous and hairy skin. Mediat. Inflamm. 2014, 2014, 105950. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Marone, G.; Granata, F.; Borriello, F.; Marone, G. Controversial role of mast cells in skin cancers. Exp. Dermatol. 2017, 26, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Rajabi, P.; Bagheri, A.; Hani, M. Intratumoral and Peritumoral Mast Cells in Malignant Melanoma: An Immunohistochemical Study. Adv. Biomed. Res. 2017, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, S.R.; Tampa, M.; Mitran, C.I.; Mitran, M.I.; Caruntu, C.; Caruntu, A.; Lupu, M.; Matei, C.; Constantin, C.; Neagu, M. Tumour Microenvironment in Skin Carcinogenesis. Adv. Exp. Med. Biol. 2020, 1226, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Harvima, I.T. Mast cell-neural interactions contribute to pain and itch. Immunol. Rev. 2018, 282, 168–187. [Google Scholar] [CrossRef]
- Vukman, K.V.; Försönits, A.; Oszvald, Á.; Tóth, E.; Buzás, E.I. Mast cell secretome: Soluble and vesicular components. Semin. Cell Dev. Biol. 2017, 67, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Duncan, L.M.; Richards, L.A.; Mihm, M.C., Jr. Increased mast cell density in invasive melanoma. J. Cutan. Pathol. 1998, 25, 11–15. [Google Scholar] [CrossRef]
- Kessler, D.A.; Langer, R.S.; Pless, N.A.; Folkman, J. Mast cells and tumor angiogenesis. Int. J. Cancer 1976, 18, 703–709. [Google Scholar] [CrossRef]
- Ribatti, D.; Ennas, M.G.; Vacca, A.; Ferreli, F.; Nico, B.; Orru, S.; Sirigu, P. Tumor vascularity and tryptase-positive mast cells correlate with a poor prognosis in melanoma. Eur. J. Clin. Investig. 2003, 33, 420–425. [Google Scholar] [CrossRef]
- Ugurel, S.; Rappl, G.; Tilgen, W.; Reinhold, U. Increased serum concentration of angiogenic factors in malignant melanoma patients correlates with tumor progression and survival. J. Clin. Oncol. 2001, 19, 577–583. [Google Scholar] [CrossRef]
- Schadendorf, D.; Möller, A.; Algermissen, B.; Worm, M.; Sticherling, M.; Czarnetzki, B.M. IL-8 produced by human malignant melanoma cells in vitro is an essential autocrine growth factor. J. Immunol. 1993, 151, 2667–2675. [Google Scholar] [PubMed]
- Halaban, R.; Kwon, B.S.; Ghosh, S.; Delli Bovi, P.; Baird, A. bFGF as an autocrine growth factor for human melanomas. Oncogene Res. 1988, 3, 177–186. [Google Scholar]
- Fisher, M.S.; Kripke, M.L. Systemic alteration induced in mice by ultraviolet light irradiation and its relationship to ultraviolet carcinogenesis. Proc. Natl. Acad. Sci. USA 1977, 74, 1688–1692. [Google Scholar] [CrossRef] [Green Version]
- Ch’ng, S.; Wallis, R.A.; Yuan, L.; Davis, P.F.; Tan, S.T. Mast cells and cutaneous malignancies. Mod. Pathol. 2006, 19, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Grimbaldeston, M.A.; Skov, L.; Finlay-Jones, J.J.; Hart, P.H. Increased dermal mast cell prevalence and susceptibility to development of basal cell carcinoma in humans. Methods 2002, 28, 90–96. [Google Scholar] [CrossRef]
- Wille, J.J.; Kydonieus, A.F.; Murphy, G.F. cis-urocanic acid induces mast cell degranulation and release of preformed TNF-alpha: A possible mechanism linking UVB and cis-urocanic acid to immunosuppression of contact hypersensitivity. Ski. Pharmacol. Appl. Ski. Physiol. 1999, 12, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, J.J.; Askenase, P.W. The effect of histamine, antihistamines, and a mast cell stabilizer on the growth of cloudman melanoma cells in DBA/2 mice. J. Investig. Dermatol. 1983, 81, 28–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, K.; Gilbertz, K.P.; Meineke, V. Serotonin and ionizing radiation synergistically affect proliferation and adhesion molecule expression of malignant melanoma cells. J. Dermatol. Sci. 2012, 68, 89–98. [Google Scholar] [CrossRef]
- Yarlagadda, K.; Hassani, J.; Foote, I.P.; Markowitz, J. The role of nitric oxide in melanoma. Biochim. Biophys. Acta. Rev. Cancer 2017, 1868, 500–509. [Google Scholar] [CrossRef]
- Mattila, J.T.; Thomas, A.C. Nitric oxide synthase: Non-canonical expression patterns. Front. Immunol. 2014, 5, 478. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef]
- Murata, J.; Tada, M.; Iggo, R.D.; Sawamura, Y.; Shinohe, Y.; Abe, H. Nitric oxide as a carcinogen: Analysis by yeast functional assay of inactivating p53 mutations induced by nitric oxide. Mutat. Res. 1997, 379, 211–218. [Google Scholar] [CrossRef]
- Lala, P.K.; Chakraborty, C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001, 2, 149–156. [Google Scholar] [CrossRef]
- Broholm, H.; Rubin, I.; Kruse, A.; Braendstrup, O.; Schmidt, K.; Skriver, E.B.; Lauritzen, M. Nitric oxide synthase expression and enzymatic activity in human brain tumors. Clin. Neuropathol. 2003, 22, 273–281. [Google Scholar] [PubMed]
- Massi, D.; De Nisi, M.C.; Franchi, A.; Mourmouras, V.; Baroni, G.; Panelos, J.; Santucci, M.; Miracco, C. Inducible nitric oxide synthase expression in melanoma: Implications in lymphangiogenesis. Mod. Pathol. 2009, 22, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, B.; Van Den Oord, J.J. Expression of the inducible isoform of nitric oxide synthase in pigment cell lesions of the skin. Br. J. Dermatol. 2000, 142, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Ye, S.; Li, K.; Huang, M.; Wang, Q.; Zeng, S.; Chen, X.; Gao, W.; Chen, J.; Zhang, Q.; et al. NOS1 inhibits the interferon response of cancer cells by S-nitrosylation of HDAC2. J. Exp. Clin. Cancer Res. 2019, 38, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, F.H.; Molognoni, F.; Morais, A.S.; Toricelli, M.; Mouro, M.G.; Higa, E.M.; Lopes, J.D.; Jasiulionis, M.G. Endothelial nitric oxide synthase uncoupling as a key mediator of melanocyte malignant transformation associated with sustained stress conditions. Free Radic. Biol. Med. 2011, 50, 1263–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahdenranta, J.; Hagendoorn, J.; Padera, T.P.; Hoshida, T.; Nelson, G.; Kashiwagi, S.; Jain, R.K.; Fukumura, D. Endothelial nitric oxide synthase mediates lymphangiogenesis and lymphatic metastasis. Cancer Res. 2009, 69, 2801–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, A.; Palma, G.; Rosati, A.; Giudice, A.; Falco, A.; Petrillo, A.; Petrillo, M.; Bimonte, S.; Di Benedetto, M.; Esposito, G.; et al. Role of endothelial nitric oxide synthase (eNOS) in chronic stress-promoted tumour growth. J. Cell. Mol. Med. 2012, 16, 920–926. [Google Scholar] [CrossRef]
- Tu, Y.T.; Tao, J.; Liu, Y.Q.; Li, Y.; Huang, C.Z.; Zhang, X.B.; Lin, Y. Expression of endothelial nitric oxide synthase and vascular endothelial growth factor in human malignant melanoma and their relation to angiogenesis. Clin. Exp. Dermatol. 2006, 31, 413–418. [Google Scholar] [CrossRef]
- Shi, Q.; Xiong, Q.; Wang, B.; Le, X.; Khan, N.A.; Xie, K. Influence of nitric oxide synthase II gene disruption on tumor growth and metastasis. Cancer Res. 2000, 60, 2579–2583. [Google Scholar]
- Garg, S.K.; Ott, M.J.; Mostofa, A.G.M.; Chen, Z.; Chen, Y.A.; Kroeger, J.; Cao, B.; Mailloux, A.W.; Agrawal, A.; Schaible, B.J.; et al. Multi-Dimensional Flow Cytometry Analyses Reveal a Dichotomous Role for Nitric Oxide in Melanoma Patients Receiving Immunotherapy. Front. Immunol. 2020, 11, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Yang, S.; Misner, B.J.; Chiu, R.; Liu, F.; Meyskens, F.L., Jr. Nitric oxide initiates progression of human melanoma via a feedback loop mediated by apurinic/apyrimidinic endonuclease-1/redox factor-1, which is inhibited by resveratrol. Mol. Cancer Ther. 2008, 7, 3751–3760. [Google Scholar] [CrossRef] [Green Version]
- Kashiwagi, S.; Izumi, Y.; Gohongi, T.; Demou, Z.N.; Xu, L.; Huang, P.L.; Buerk, D.G.; Munn, L.L.; Jain, R.K.; Fukumura, D. NO mediates mural cell recruitment and vessel morphogenesis in murine melanomas and tissue-engineered blood vessels. J. Clin. Investig. 2005, 115, 1816–1827. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Tu, Y.T.; Li, J.W.; Feng, A.P.; Huang, C.Z.; Wu, Y.; Shen, G.X. Endogenous production of nitric oxide contributes to proliferation effect of vascular endothelial growth factor-induced malignant melanoma cell. Clin. Exp. Dermatol. 2006, 31, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Fraix, A.; Conte, C.; Gazzano, E.; Riganti, C.; Quaglia, F.; Sortino, S. Overcoming Doxorubicin Resistance with Lipid-Polymer Hybrid Nanoparticles Photoreleasing Nitric Oxide. Mol. Pharm. 2020, 17, 2135–2144. [Google Scholar] [CrossRef]
- Paskaš, S.; Krajnović, T.; Basile, M.S.; Dunđerović, D.; Cavalli, E.; Mangano, K.; Mammana, S.; Al-Abed, Y.; Nicoletti, F.; Mijatović, S.; et al. Senescence as a main mechanism of Ritonavir and Ritonavir-NO action against melanoma. Mol. Carcinog. 2019, 58, 1362–1375. [Google Scholar] [CrossRef]
- Paskas, S.; Mazzon, E.; Basile, M.S.; Cavalli, E.; Al-Abed, Y.; He, M.; Rakocevic, S.; Nicoletti, F.; Mijatovic, S.; Maksimovic-Ivanic, D. Lopinavir-NO, a nitric oxide-releasing HIV protease inhibitor, suppresses the growth of melanoma cells in vitro and in vivo. Investig. New Drugs 2019, 37, 1014–1028. [Google Scholar] [CrossRef] [PubMed]
- Kaoud, T.S.; Mohassab, A.M.; Hassan, H.A.; Yan, C.; Van Ravenstein, S.X.; Abdelhamid, D.; Dalby, K.N.; Abdel-Aziz, M. NO-releasing STAT3 inhibitors suppress BRAF-mutant melanoma growth. Eur. J. Med. Chem. 2020, 186, 111885. [Google Scholar] [CrossRef]
- Zang, Y.; Lai, F.; Fu, J.; Li, C.; Ma, J.; Chen, C.; Liu, K.; Zhang, T.; Chen, X.; Zhang, D. Novel nitric oxide-releasing derivatives of triptolide as antitumor and anti-inflammatory agents: Design, synthesis, biological evaluation, and nitric oxide release studies. Eur. J. Med. Chem. 2020, 190, 112079. [Google Scholar] [CrossRef]
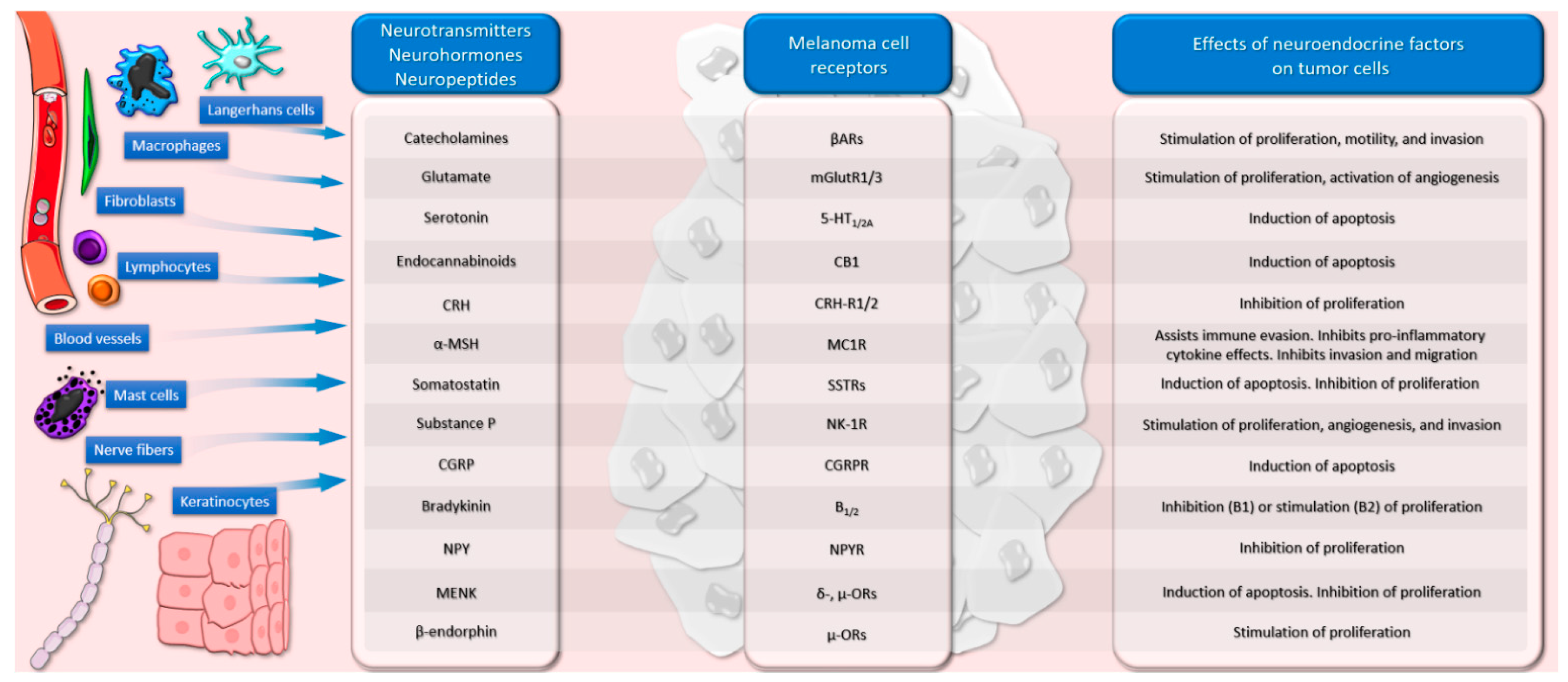

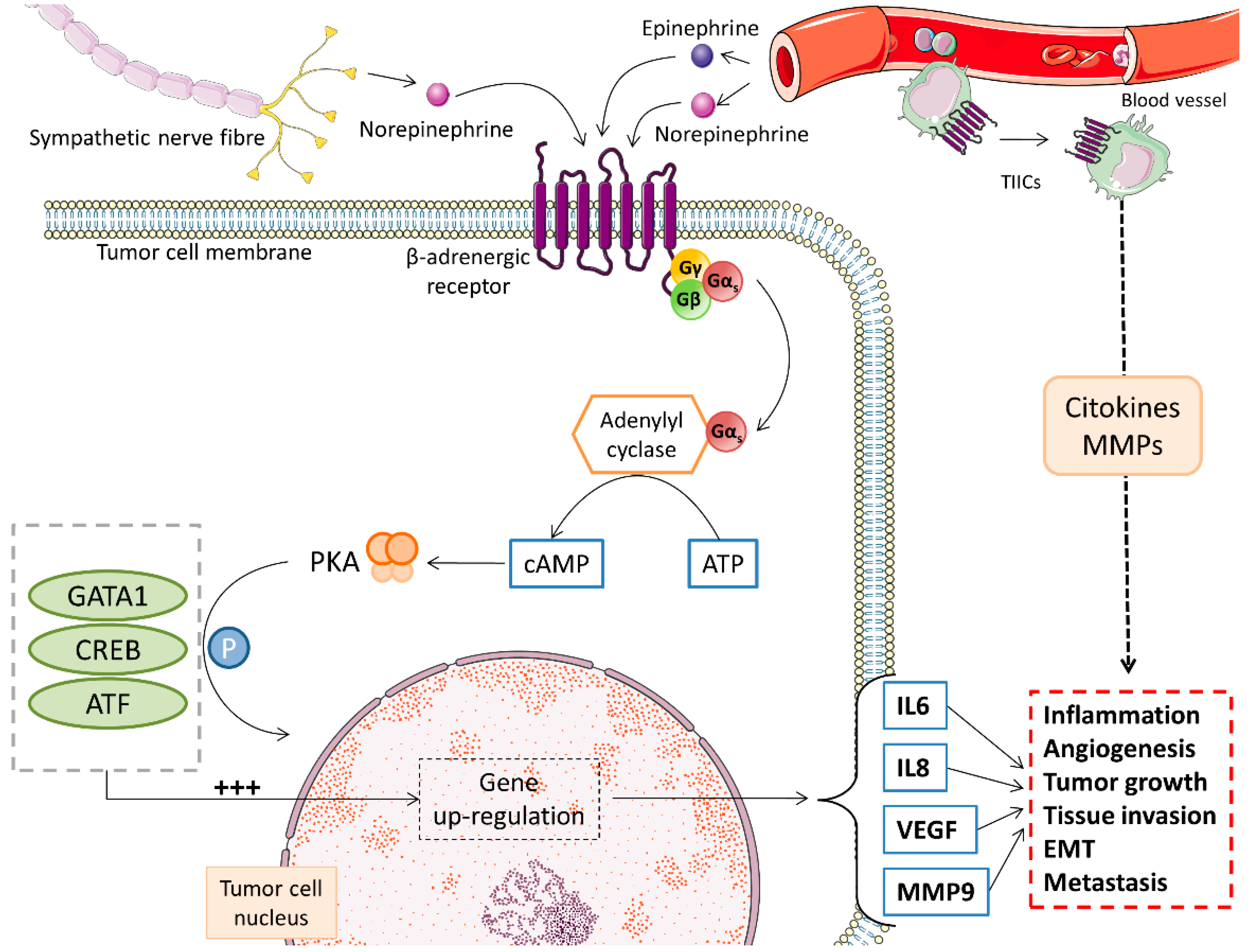

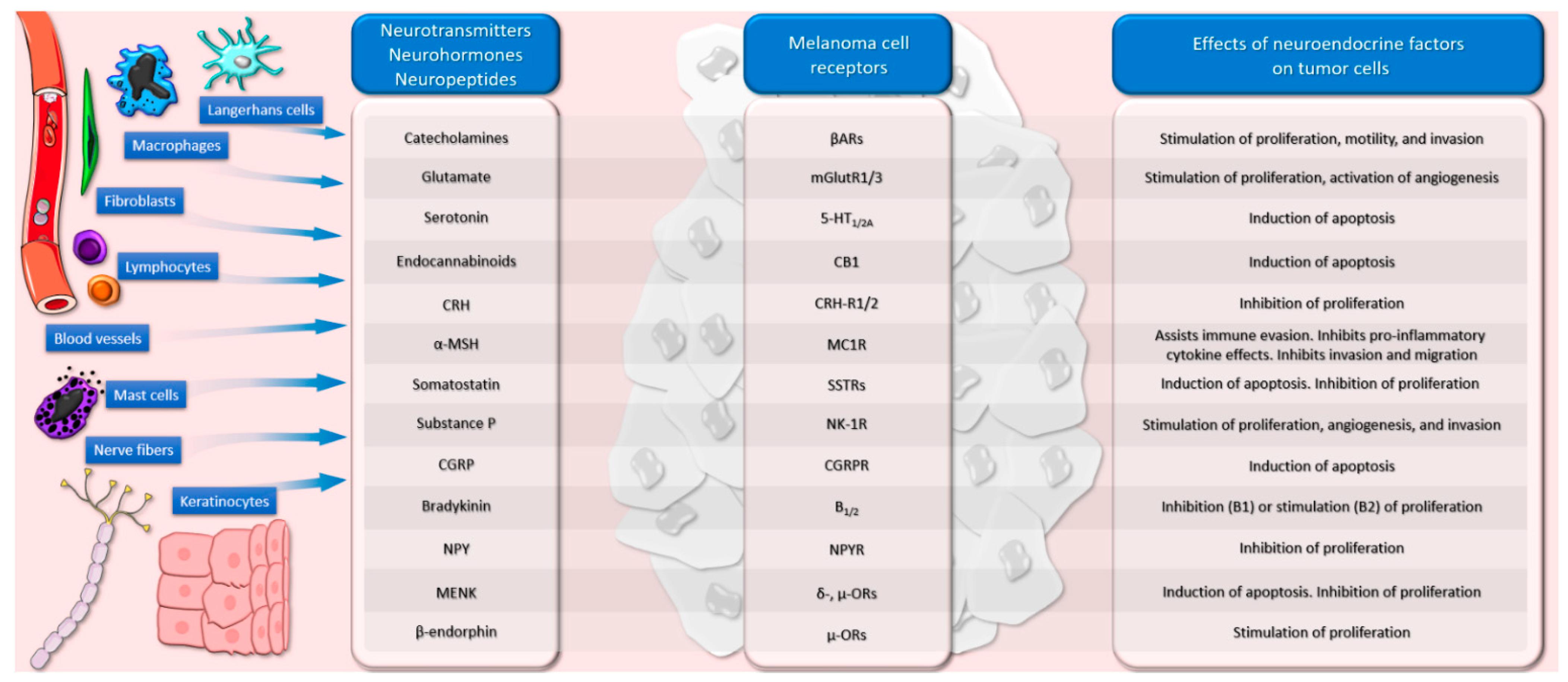{kind=link}
{kind=link}
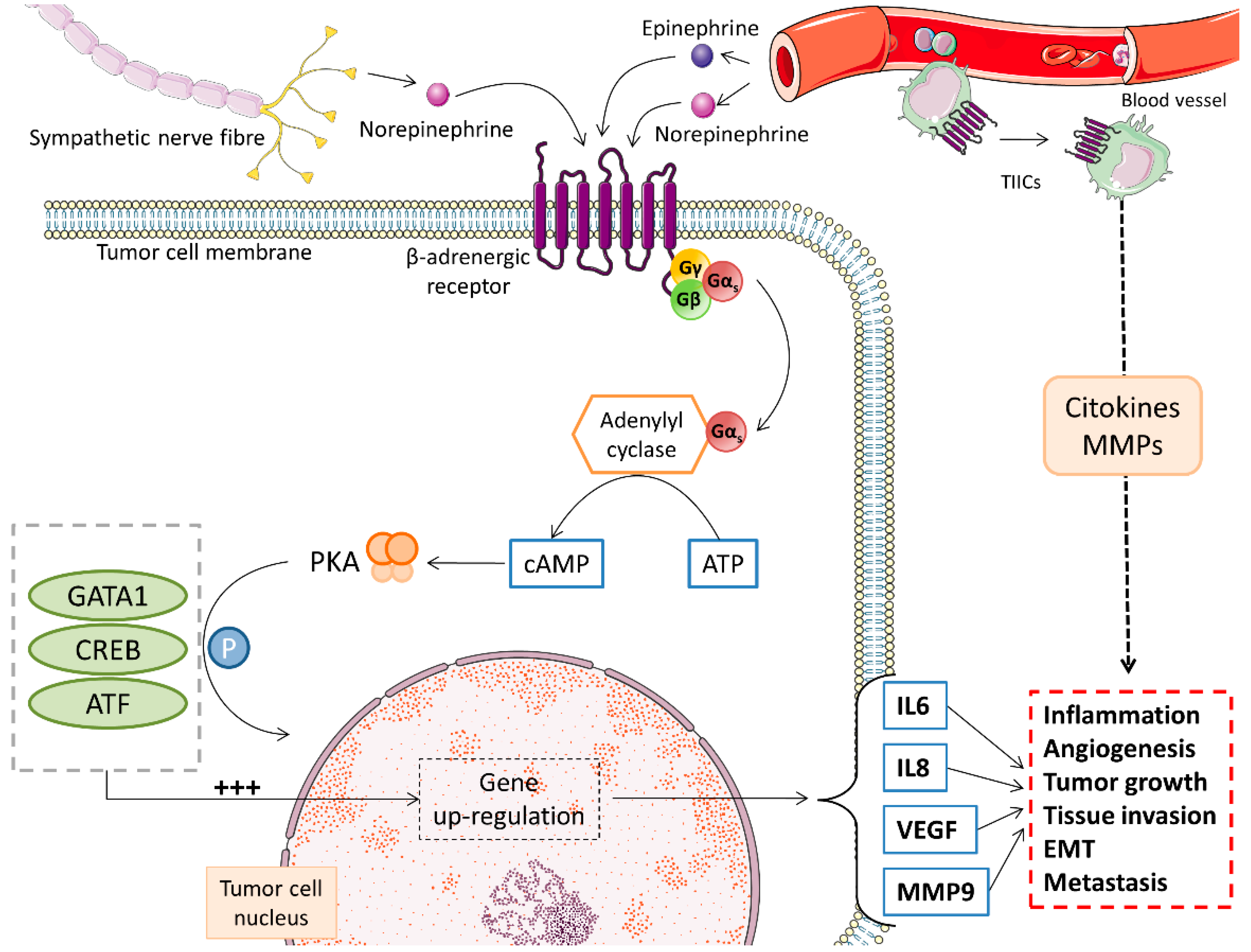{kind=link}
| Factor | Experimental Model | Mechanism | Effect | Reference |
|---|---|---|---|---|
| Catecholamines | ||||
| Norepinephrine | In vitro (C8161, 1174MEL, and Me18105) | Release of VEGF, IL-6, IL-8 | Stimulation of melanoma aggressiveness | [38] |
| In vitro (A375 cells) | Activation of β3-ARs | Recruitment of stromal cells | [73] | |
| Norepinephrine, epinephrine | In vitro (B16F10 melanoma cells) | β-Ars 1-mediated effects | Stimulation of cell proliferation | [17] |
| In vitro (Hs29-4T and A375 cells) | Activation of MAPKs and MMPs 2 and 9 | Stimulation of motility and invasion | [58] | |
| Epinephrine | In vitro (FM-55-P, 92-1, Mel202, and A375 cells) | β2-ARs-mediated effects | Stimulation of cell proliferation and invasion | [59] |
| Epinephrine | In vivo (mice with B16F10 melanoma inoculations) | Mobilization of NK cells and redistribution to tumors in regular exercise | Inhibition of tumor incidence and growth | [96] |
| Phenylephrine | In vitro (SK-Mel 23 cells) | p38 and ERKs signaling via α1-ARs stimulation | Inhibition of cell proliferation | [97] |
| Glutamate | ||||
| Glutamate | In vivo (mice with C8161 xenografts) | Activation of mGlut1 | Stimulation of cell proliferation and metabolic activity | [105] |
| In vivo (mice with UACC903-G4 xenografts) | Activation of PI3K/Akt/mTOR pathway | Stimulation of angiogenesis in xenografts | [110] | |
| Serotonin and Analogs | ||||
| Serotonin | In vitro (B16F10 melanoma cells) | 5-HT2A-mediated cross-talk with SP | Neutralization of the apoptotic effects of SP | [123] |
| Serotonin | In vitro (B16F10, SK-MEL-2, and Melan-A cells) | 5-HT2A-mediated increase in tyrosinase activity, dendritic network, and melanin production | Induction of melanogenesis | [124] |
| 1-NPZ 2 | In vitro (MNT-1 melanoma cells) | Increased expression of Cox-2 and ROS production. | Induction of apoptosis | [130] |
| NCS, NPCS 3 | In vitro (murine B16 and human HMV-II melanoma cells) | Inhibition of tyrosinase | Suppression of melanogenesis | [131] |
| Tegaserod | In vivo (mice with B16F10 melanoma inoculations) | Inhibition of PI3K/Akt/mTOR pathway | Inhibition of tumor growth and dissemination. Increase in survival | [134] |
| Cannabinoids | ||||
| Anandamide | In vitro (A375 melanoma cells) | CB1-mediated induction of caspase-dependent apoptosis; Cox-2 and Lox-mediated cytotoxicity | Induction of apoptosis | [143] |
| THC, WIN 55,212-2 | In vitro (B16 and A375 melanoma cells) | CB1/2-mediated inhibition of Akt signaling inhibition | Induction of apoptosis. Inhibition of cell proliferation and metastasis | [145] |
| Anandamide, ACEA 4 | In vitro (HT168-M1 and WM983B melanoma cells) | CB1-mediated inhibition of PI3K/Akt/mTOR pathway | Inhibition of cell proliferation and metastasis | [147] |
| Cannabidiol | In vivo (mice with B16F10 melanoma inoculations) | Possibly CB2-mediated antitumoral activity | Inhibition of tumor growth. Increased survival and quality of life | [149] |
| WIN 55,212-2 | In vitro (COLO38, SKMEL28, and OCM1A melanoma cells) | Lipid raft-mediated activation of capsase-9 and ERK pathways | Inhibition of cell proliferation | [150] |
| THC, Cannabidiol | In vitro (CHL-1, A375, and SK-MEL-28 melanoma cells) | Atg7-mediated autophagy; TRIB3-mediated apoptosis | Inhibition of cell viability | [151] |
| Cannabis | Human retrospective study | Untested, possibly PD-1 or PD-L1 inhibition | Decrease of immunotherapy (nivolumab) response rate | [153] |
| Factor | Experimental Model | Mechanism | Effect | Reference |
|---|---|---|---|---|
| CRH-POMC | ||||
| CRH | In vitro (B16F0 and B16F10) | Activation of ERK1/2 pathways | Stimulation of tumor cell migration | [160] |
| CRH and various analogs | In vitro (murine Cloudman and B16 tumors) | CRH1-mediated Ca2+ signaling alteration | Inhibition of cell proliferation | [161] |
| POMC | In vivo (mice with B16F10 melanoma inoculations) | Induction of differentiation. Direct effect on endothelial cells. | Inhibition of cell proliferation and angiogenesis. Increased survival | [163] |
| Autophagy and apoptosis via α-MSH/HIF-1α/ BNIP3 signaling pathway | Induction of apoptosis | [164] | ||
| Alpha-MSH | ||||
| α-MSH | In vitro (B16BL6 and B16F1 cells) | Inhibition of MMPs 2 and 9 | Inhibition of invasion and migration | [179] |
| In vitro (A-375SM and HBL melanoma cells) | Reduction of NF-κB DNA activation | Immunomodulation. Inhibition of proinflammatory cytokine effects | [180] | |
| In vitro (STG, RIDE, WR2 cultures, HBL, DOR cells) | MC1R-mediated reduction of ICAM-1 expression | Inhibition of proinflammatory cytokine effects | [183,184] | |
| In vitro (HBL and A375-SM melanoma cells) | MC1R-mediated elevation of cAMP and intracellular Ca2+ | Inhibition of invasion | [187] | |
| In vitro (HBL melanoma cells) | Decreased integrin expression | Inhibition of invasion | [188] | |
| In vitro (SOM, 177w7B7, and VUP cell lines) | Modulation of tumor microenvironment. Opposing effects to TNF-α | Inhibition of invasion | [189] | |
| In vitro (A375-SM and HBL cells) | Decrease in B7 expression | Assists melanoma cells in immune evasion | [200] | |
| Inhibition of NF-κB binding activity | Assists melanoma cells in immune evasion | [201] | ||
| TRH | ||||
| TRH | In vitro (COS and B16 melanoma cells) | Stimulation of cAMP production | Similar MC1R activation to α-MSH | [206] |
| Somatostatin Analogs | ||||
| Octreotide and SOM230 | In vitro (18 human melanoma cell lines) | Receptor-mediated mechanism (mainly SSTR-2) | Inhibition of cell proliferation | [211] |
| PSM 1 | In vivo (mice with B16F10 melanoma inoculations) | Calreticulin exposure. Decreased expression of Ki67. Modulation of EMT markers expression. Increased TNF-α, IFN-γ, and IL-2 in T cells | Induction of apoptosis. Inhibition of cell proliferation and invasion | [212] |
| Pasireotide | Patients with advanced melanoma (n = 10) | Inhibition of Ki-67 expression and serum levels of IGF-1, IGF-2, MIA, S100B, and IGFBP3 | Partial response (n = 1)/stable disease (n = 1) | [213] |
| Vasopressin Analogs | ||||
| Desmopressin | In vivo (mice with B16F10 melanoma inoculations) | TIMP-1-dependent prevention of melanoma cells implantation | Inhibition of dissemination | [220] |
| Factor | Experimental Model | Mechanism | Effect | Reference |
|---|---|---|---|---|
| Substance P/NK-1R Antagonists | ||||
| Substance P | In vitro (B16F10 cells) | NK-1R-mediated inhibition of MAPK and decrease in tyrosinase activity | Inhibition of melanogenesis | [234] |
| Maropitant | In vitro, in vivo (canine melanoma tissue/cells) | Possible NK-1R-mediated effects | Inhibition of cell proliferation. Induction of apoptosis | [224] |
| Aprepitant | In vitro (COLO 858, MEL HO, and COLO 679 cells) | Inhibition of SP-induced mitogen stimulation | Inhibition of cell proliferation | [225] |
| L732138 | In vitro (A375 and B16F10 cells) | Inhibition of MMP-2 and -9 expression via ERK1/2, JNK, and p38 signaling | Inhibition of dissemination | [226] |
| L-733 060 | In vitro (COLO 858, MEL HO, and COLO 679 cells) | Inhibition of mitogenesis via MAPK pathway | Inhibition of cell proliferation | [228] |
| Cyclosporin A | In vitro (COLO 858, MEL HO, and COLO 679 cells) | Inhibition of SP-induced mitogen stimulation | Inhibition of cell proliferation | [229] |
| AA3266 | In vitro (MeW164, MeW155, MeW151 cells) | Decreased Ki-67 expression. Induction of cell cycle arrest | Inhibition of cell proliferation | [232] |
| CGRP | ||||
| CGRP | In vitro (B16F10 cells) | Suppression of NF-κB activation. Promotion of apoptosis | Inhibition of cell proliferation | [238] |
| Bradykinin | ||||
| Bradykinin | In vitro (B16-BL6 cells) | B2-mediated increased expression of endothelin-1 | Stimulation of cell proliferation | [246] |
| DABK | In vivo (murine Tm5 melanoma) | B1-mediated increase in Ca2+ concentration and ERK phosphorylation | Inhibition of cell proliferation, invasion, dissemination, and vascularization | [249,250] |
| Neuropeptide Y Analogs | ||||
| BIIE 0246 | In vivo (mice with B16F10 melanoma inoculations) | NPY Y2-R antagonism. Decrease in VEGF serum levels | Inhibition of cell proliferation and angiogenesis | [254] |
| GRP | ||||
| GRP | In vivo (mice with B16F10 melanoma inoculations) | Antibodies induced via a multicomponent vaccine formulation | Inhibition of cell proliferation | [269] |
| Enkephalins | ||||
| MENK | In vivo (mice with B16F10 melanoma inoculations) | Stimulation of host immune response. Direct cytotoxicity. Modulation of tumoral milieu | Inhibition of cell proliferation | [283] |
| Induction of cell cycle arrest. Increased TNF-α, IFN-γ, and IL-2 | Inhibition of cell proliferation. Increased survival | [284] | ||
| In vitro (A375 cells) | Induction of cell cycle arrest. Decreased expression of survivin | Inhibition of cell proliferation. Induction of apoptosis | [285] | |
| Beta-Endorphin | ||||
| β-endorphin | In vivo (mice with B16F10 melanoma inoculations) | Inhibition of tumor infiltration by lymphocytes | Stimulates cell proliferation. Inhibition of the host immune response | [305] |
| Vasoactive Intestinal Peptide | ||||
| VIP | In vitro (B16F10 cells) | PKA-CREB-mediated increase in tyrosinase activity | Induction of melanogenesis | [309] |
| Factor | Experimental Model | Mechanism | Effect | Reference |
|---|---|---|---|---|
| iNOS | Ex vivo (various human melanoma tissue specimens) | Stimulation of blood and lymph angiogenesis | Stimulation of blood and lymphatic dissemination through angiogenesis | [335] |
| In vivo (mice with B16BL6 melanoma inoculations) | Modulation of NO-sensitive macrophages’ activity | Stimulation of cell proliferation and dissemination | [342] | |
| nNOS | In vitro (A375 cells) In vivo (mice with B16F10 melanoma inoculations) | Inhibition of IFNα signaling and tumor infiltration by lymphocytes | Stimulation of dissemination | [337] |
| uncoupled eNOS | In vitro (Tm5 melanoma cells) | Production of superoxide | Stimulation of cell proliferation and malignant transformation | [338] |
| eNOS | In vivo (mice with B16F10 melanoma inoculations) | Stimulation of peritumor lymphatic hyperplasia via PI3K pathway activation | Stimulation of cell proliferation and dissemination (lymphatic) | [339] |
| β-adrenergic stimulation of cAMP-PKA signaling | Stimulation of cell proliferation in stress conditions | [340] | ||
| Ex vivo (melanoma tissue sections) | VEGF-mediated increase in microvascular density | Stimulation of angiogenesis | [341] | |
| NO | Ex vivo (advanced melanoma resections) | Modulation of immune cells’ activity | Increased/decreased survival dependent on NO source | [343] |
| In vitro (Lu1205 metastatic melanoma cells) | APE/Ref-1-mediated activation of oncogenic targets | Stimulation of cell proliferation and dissemination | [344] | |
| In vivo (mice with B16F1 and B16F10 inoculations) | Direct effect on endothelial cells. Indirect effect by stimulating angiogenic factors | Stimulation of angiogenesis and dissemination | [345] | |
| In vitro (A375 cells) | VEGF-induced stimulation of iNOS expression | Stimulation of cell proliferation | [346] | |
| In vitro (M14 DOX-resistant melanoma cells) | NO delivered by nanoparticles increases DOX retention | Stimulation of the antitumoral activity of doxorubicin | [347] | |
| In vitro (metastatic B16F10 cells) | Induction of senescence. Inhibition of S6 protein | Stimulation of the antitumoral activity of Ritonavir | [348] | |
| In vitro (B16, B16F10, A375 cells) In vivo (mice with B16 melanoma inoculations) | Production of ROS and RNS. Inhibition of P70S6K | Stimulation of the antitumoral activity of Lopinavir | [349] | |
| In vitro (A375 cells) | NO delivered by synthetic quinolone donors. Inhibition of STAT3 tyrosine phosphorylation. Production of ROS. Induction of cell cycle arrest | Inhibition of cell proliferation | [350] | |
| In vivo (mice with B16F10 melanoma inoculations) | NO delivered by triptolide/furoxans hybrids | Inhibition of cell proliferation. Anti-inflammatory effects | [351] |
| Tested Substance | Study Type | Mechanism | Effect | Reference |
|---|---|---|---|---|
| β-blockers (metoprolol, propranolol, atenolol, and others) | Cohort study (4179 patients) | Blocking βARs | Increased survival in melanoma patients | [83] |
| β-blockers (unspecified) | Cohort study (121 patients) | Blocking βARs | Reduced risk of disease progression | [84] |
| β-blockers (pan or β1 selective) | Cohort study (195 patients) | Inhibition of stress signaling, particularly via β2AR signaling | Increased survival in melanoma patients | [86] |
| β-blockers (pan or β1 selective) | Cohort study (203 patients) | Blocking βARs | No impact on survival in melanoma patients | [91] |
| Riluzole | Phase 0 trial (12 patients) | Glutamate blockade inhibiting MAPK and PI3k/Akt signaling | Melanoma metabolic activity suppression was achieved. Inconsistent effects | [117] |
| Riluzole | Phase II trial (13 patients) | Inhibition of GRM1 signaling Increased leukocyte infiltration | Antitumoral biological effects. No tumoral response recorded according to staging criteria | [100] |
| Ipilimumab | Phase II trial (75 patients) | NO-mediated modulation of the tumor microenvironment | Mixed anti- and pro-melanoma activity. Inconsistent effects of NO and metabolites | [343] |
| SSRIs (unspecified) | Cohort study (5591 patients) | Inhibition of serotonin uptake | Decrease in survival of melanoma patients | [141] |
| Cannabis | Cohort study (140 patients) | Complex interaction with the immune response, possibly CB2-mediated | Decrease of response rate to nivolumab in patients with advanced melanoma | [153] |
| Pasireotide | Phase I trial (10 patients) | SSTRs-mediated Ras/MAPK signaling modulation | Stable disease and partial response were obtained. Progression of disease also recorded in some patients | [213] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheau, C.; Draghici, C.; Ilie, M.A.; Lupu, M.; Solomon, I.; Tampa, M.; Georgescu, S.R.; Caruntu, A.; Constantin, C.; Neagu, M.; et al. Neuroendocrine Factors in Melanoma Pathogenesis. Cancers 2021, 13, 2277. https://doi.org/10.3390/cancers13092277
Scheau C, Draghici C, Ilie MA, Lupu M, Solomon I, Tampa M, Georgescu SR, Caruntu A, Constantin C, Neagu M, et al. Neuroendocrine Factors in Melanoma Pathogenesis. Cancers. 2021; 13(9):2277. https://doi.org/10.3390/cancers13092277
Chicago/Turabian StyleScheau, Cristian, Carmen Draghici, Mihaela Adriana Ilie, Mihai Lupu, Iulia Solomon, Mircea Tampa, Simona Roxana Georgescu, Ana Caruntu, Carolina Constantin, Monica Neagu, and et al. 2021. "Neuroendocrine Factors in Melanoma Pathogenesis" Cancers 13, no. 9: 2277. https://doi.org/10.3390/cancers13092277
APA StyleScheau, C., Draghici, C., Ilie, M. A., Lupu, M., Solomon, I., Tampa, M., Georgescu, S. R., Caruntu, A., Constantin, C., Neagu, M., & Caruntu, C. (2021). Neuroendocrine Factors in Melanoma Pathogenesis. Cancers, 13(9), 2277. https://doi.org/10.3390/cancers13092277