
A Gene Signature Derived from the Loss of CDKN1A (p21) Is Associated with CMS4 Colorectal Cancer

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
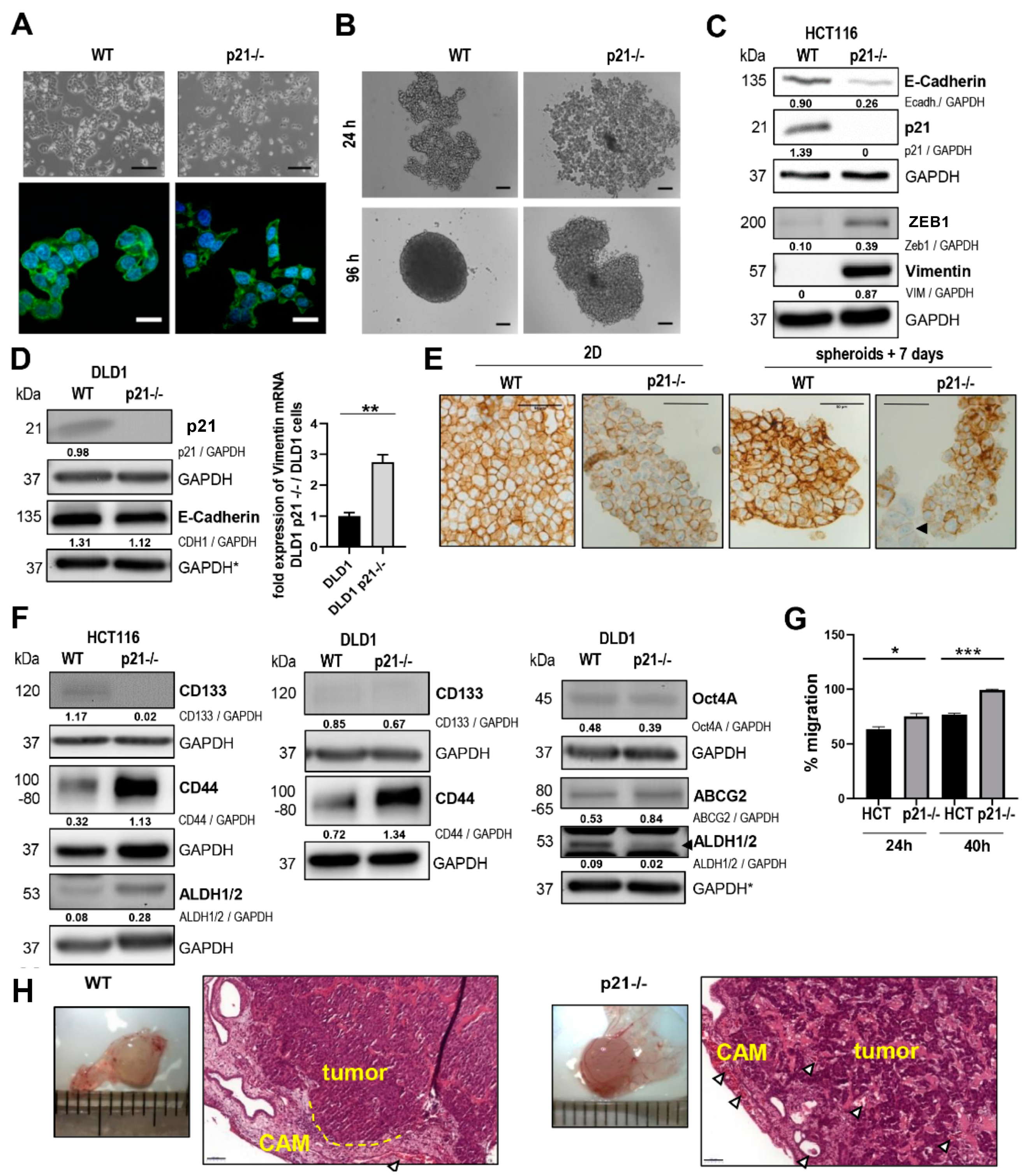
2.1. p21 Loss Induced a Mesenchymal Phenotype in HCT116 Colorectal Cancer Cells
2.2. Differential Gene Expression Signature of HCT116 p21-/- Colorectal Cancer Cells
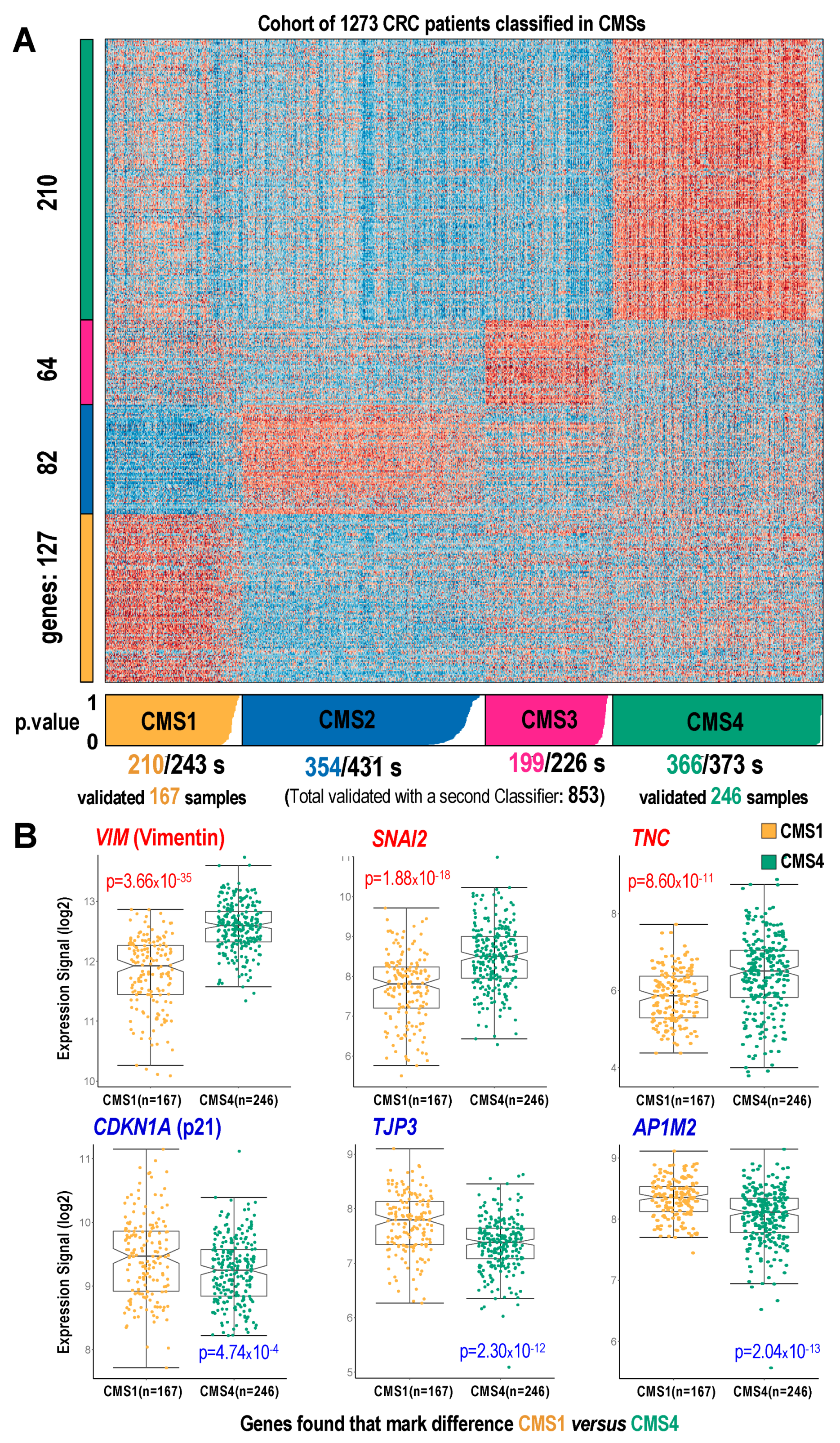
2.3. Mapping the p21-/- Gene Signature on CMS Subtypes Using a Cohort of CRC Samples
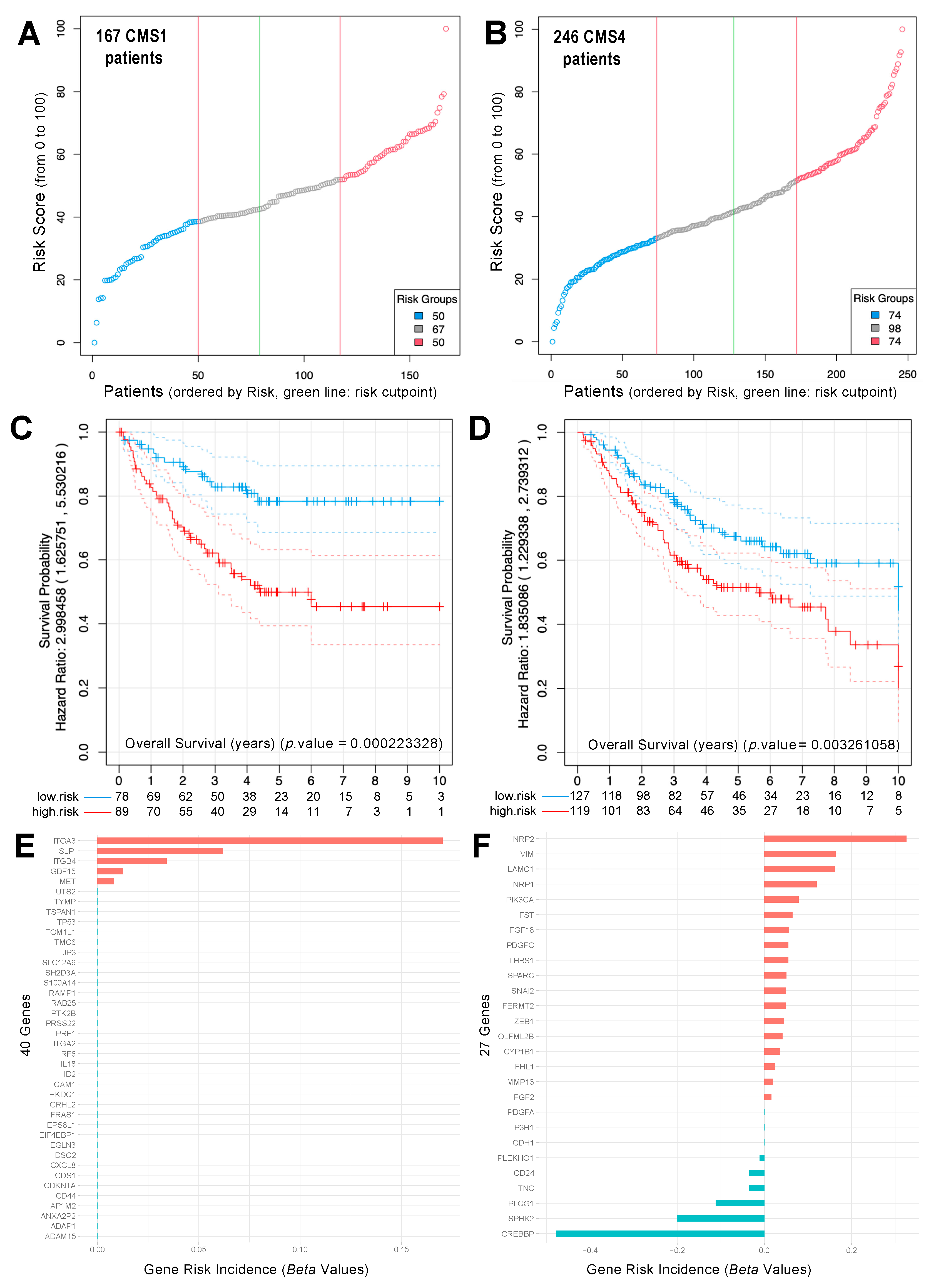
2.4. Analysis of Survival and Risk of CRC Samples Classified as CMS1 and CMS4
2.5. Functional Differences in Genes Included in CRC Subtypes CMS1 and CMS4
3. Discussion
4. Material and Methods
4.1. Human Colorectal Carcinoma Cell Lines
4.2. Morphology and Fluorescence Staining
4.3. Collection of Cell Pellets for Western Blot, RT-qPCR, and NanoString Analysis
4.4. Western Blotting
4.5. 3D Spheroid Generation and Immunostaining
4.6. Wound Healing Assay
4.7. Gene Expression Measurement with the NanoString Platform
4.8. Differential Expression Calculation and CMS Gene Signature Identification in CRC Cohort
4.9. Description of the Cohort of CRC Primary Tumor Samples Used in This Study
4.10. Colon Cancer Cohort Risk Prediction and Survival Analysis in Different CMSubtypes
4.11. Quantitative PCR (qPCR) for Gene Expression
4.12. Chorioallantoic Membrane Xenograft Assay
4.13. Plasmids and Transfection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fakih, M.G. Metastatic colorectal cancer: Current state and future directions. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1809–1824. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.; Calon, A.; Lonardo, E.; Batlle, E. Determinants of metastatic competency in colorectal cancer. Mol. Oncol. 2017, 11, 97–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, T.; Datta, P.K. Regulation of EMT in Colorectal Cancer: A Culprit in Metastasis. Cancers 2017, 9, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Hara, T.; Choi, Y.; Subramanian, M.; Francis, P.; Bilke, S.; Walker, R.L.; Pineda, M.; Zhu, Y.; Yang, Y.; et al. A p21-ZEB1 complex inhibits epithelial-mesenchymal transition through the microRNA 183-96-182 cluster. Mol. Cell. Biol. 2014, 34, 533–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, P.F.; Jiang, T.; Chen, F.; Shi, P.C.; Li, H.Q.; Bai, J.; Song, J. KIF4A facilitates cell proliferation via induction of p21-mediated cell cycle progression and promotes metastasis in colorectal cancer. Cell Death Dis. 2018, 9, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Bancroft, L.; Augenlicht, L.H. Methylation in the p21WAF1/cip1 promoter of Apc+/-, p21+/- mice and lack of response to sulindac. Oncogene 2005, 24, 2104–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Velcich, A.; Lozonschi, I.; Liang, J.; Nicholas, C.; Zhuang, M.; Bancroft, L.; Augenlicht, L.H. Inactivation of p21WAF1/cip1 enhances intestinal tumor formation in Muc2-/- mice. Am. J. Pathol. 2005, 166, 1239–1246. [Google Scholar] [CrossRef]
- Karakas, B.; Weeraratna, A.; Abukhdeir, A.; Blair, B.G.; Konishi, H.; Arena, S.; Becker, K.; Wood, W., 3rd; Argani, P.; De Marzo, A.M.; et al. Interleukin-1 alpha mediates the growth proliferative effects of transforming growth factor-beta in p21 null MCF-10A human mammary epithelial cells. Oncogene 2006, 25, 5561–5569. [Google Scholar] [CrossRef] [Green Version]
- Karakas, B.; Weeraratna, A.T.; Abukhdeir, A.M.; Konishi, H.; Gustin, J.P.; Vitolo, M.I.; Bachman, K.E.; Park, B.H. P21 gene knock down does not identify genetic effectors seen with gene knock out. Cancer Biol. Ther. 2007, 6, 1025–1030. [Google Scholar] [CrossRef] [Green Version]
- Lindner, P.; Paul, S.; Eckstein, M.; Hampel, C.; Muenzner, J.K.; Erlenbach-Wuensch, K.; Ahmed, H.P.; Mahadevan, V.; Brabletz, T.; Hartmann, A.; et al. EMT transcription factor ZEB1 alters the epigenetic landscape of colorectal cancer cells. Cell Death Dis. 2020, 11, 147. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Menter, D.G.; Davis, J.S.; Broom, B.M.; Overman, M.J.; Morris, J.; Kopetz, S. Back to the Colorectal Cancer Consensus Molecular Subtype Future. Curr. Gastroenterol. Rep. 2019, 21, 5. [Google Scholar] [CrossRef] [PubMed]
- Trinh, A.; Ladrach, C.; Dawson, H.E.; Ten Hoorn, S.; Kuppen, P.J.K.; Reimers, M.S.; Koopman, M.; Punt, C.J.A.; Lugli, A.; Vermeulen, L.; et al. Tumour budding is associated with the mesenchymal colon cancer subtype and RAS/RAF mutations: A study of 1320 colorectal cancers with Consensus Molecular Subgroup (CMS) data. Br. J. Cancer 2018, 119, 1244–1251. [Google Scholar] [CrossRef] [Green Version]
- Linnekamp, J.F.; Hooff, S.R.V.; Prasetyanti, P.R.; Kandimalla, R.; Buikhuisen, J.Y.; Fessler, E.; Ramesh, P.; Lee, K.; Bochove, G.G.W.; de Jong, J.H.; et al. Consensus molecular subtypes of colorectal cancer are recapitulated in in vitro and in vivo models. Cell Death Differ. 2018, 25, 616–633. [Google Scholar] [CrossRef] [PubMed]
- Fontanillo, C.; Nogales-Cadenas, R.; Pascual-Montano, A.; De las Rivas, J. Functional analysis beyond enrichment: Non-redundant reciprocal linkage of genes and biological terms. PLoS ONE 2011, 6, e24289. [Google Scholar] [CrossRef] [Green Version]
- Roepman, P.; Schlicker, A.; Tabernero, J.; Majewski, I.; Tian, S.; Moreno, V.; Snel, M.H.; Chresta, C.M.; Rosenberg, R.; Nitsche, U.; et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int. J. Cancer 2014, 134, 552–562. [Google Scholar] [CrossRef]
- Martinez-Romero, J.; Bueno-Fortes, S.; Martin-Merino, M.; Ramirez de Molina, A.; De Las Rivas, J. Survival marker genes of colorectal cancer derived from consistent transcriptomic profiling. BMC Genom. 2018, 19, 857. [Google Scholar] [CrossRef] [Green Version]
- Eide, P.W.; Bruun, J.; Lothe, R.A.; Sveen, A. CMScaller: An R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci. Rep. 2017, 7, 16618. [Google Scholar] [CrossRef] [Green Version]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Merlos-Suarez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Cespedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Munoz, P.; et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fessler, E.; Drost, J.; van Hooff, S.R.; Linnekamp, J.F.; Wang, X.; Jansen, M.; De Sousa, E.M.F.; Prasetyanti, P.R.; JE, I.J.; Franitza, M.; et al. TGFbeta signaling directs serrated adenomas to the mesenchymal colorectal cancer subtype. EMBO Mol. Med. 2016, 8, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Wang, H.; Horbinski, C.; Wu, H.; Liu, Y.; Sheng, S.; Liu, J.; Weiss, H.; Stromberg, A.J.; Wang, C. NanoStringDiff: A novel statistical method for differential expression analysis based on NanoString nCounter data. Nucleic Acids Res. 2016, 44, e151. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Xu, E.; Liu, H.; Wan, L.; Lai, M. Epithelial-mesenchymal transition in colorectal cancer metastasis: A system review. Pathol. Res. Pr. 2015, 211, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Komor, M.A.; Bosch, L.J.; Bounova, G.; Bolijn, A.S.; Delis-van Diemen, P.M.; Rausch, C.; Hoogstrate, Y.; Stubbs, A.P.; de Jong, M.; Jenster, G.; et al. Consensus molecular subtype classification of colorectal adenomas. J. Pathol. 2018, 246, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Cybulska, M.; Olesinski, T.; Goryca, K.; Paczkowska, K.; Statkiewicz, M.; Kopczynski, M.; Grochowska, A.; Unrug-Bielawska, K.; Tyl-Bielicka, A.; Gajewska, M.; et al. Challenges in Stratifying the Molecular Variability of Patient-Derived Colon Tumor Xenografts. BioMed Res. Int. 2018, 2018, 2954208. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.F.; Chen, M.; Wu, Y.; Chen, M.N.; Glogowska, A.; Klonisch, T.; Zhang, G.J. Inhibitor of DNA Binding 2 Inhibits Epithelial-Mesenchymal Transition via Up-Regulation of Notch3 in Breast Cancer. Transl. Oncol. 2018, 11, 1259–1270. [Google Scholar] [CrossRef]
- Fontana, E.; Eason, K.; Cervantes, A.; Salazar, R.; Sadanandam, A. Context matters-consensus molecular subtypes of colorectal cancer as biomarkers for clinical trials. Ann. Oncol. 2019, 30, 520–527. [Google Scholar] [CrossRef]
- Meyer, S.N.; Galvan, J.A.; Zahnd, S.; Sokol, L.; Dawson, H.; Lugli, A.; Zlobec, I. Co-expression of cytokeratin and vimentin in colorectal cancer highlights a subset of tumor buds and an atypical cancer-associated stroma. Hum. Pathol. 2019, 87, 18–27. [Google Scholar] [CrossRef]
- Saxena, K.; Jolly, M.K.; Balamurugan, K. Hypoxia, partial EMT and collective migration: Emerging culprits in metastasis. Transl. Oncol. 2020, 13, 100845. [Google Scholar] [CrossRef]
- Amrutha, M.; Reshma, R.R.; Gayathri, M.; Padmaja, K.P.; Tessy, T.M. Reporters of Cancer Stem Cells as a Tool for Drug Discovery. Front. Oncol. 2021, 11, 669250. [Google Scholar] [CrossRef]
- Kong, D.; Zhou, H.; Neelakantan, D.; Hughes, C.J.; Hsu, J.Y.; Srinivasan, R.R.; Lewis, M.T.; Ford, H.L. VEGF-C mediates tumor growth and metastasis through promoting EMT-epithelial breast cancer cell crosstalk. Oncogene 2021, 40, 964–979. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Willis, J.A.; Reumers, J.; Taggart, M.W.; San Lucas, F.A.; Thirumurthi, S.; Kanth, P.; Delker, D.A.; Hagedorn, C.H.; Lynch, P.M.; et al. Colorectal premalignancy is associated with consensus molecular subtypes 1 and 2. Ann. Oncol. 2018, 29, 2061–2067. [Google Scholar] [CrossRef]
- Polyak, K.; Waldman, T.; He, T.C.; Kinzler, K.W.; Vogelstein, B. Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev. 1996, 10, 1945–1952. [Google Scholar] [CrossRef] [Green Version]
- Gartel, A.L. p21(WAF1/CIP1) and cancer: A shifting paradigm? BioFactors 2009, 35, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Muenzner, J.K.; Kunze, P.; Lindner, P.; Polaschek, S.; Menke, K.; Eckstein, M.; Geppert, C.I.; Chanvorachote, P.; Baeuerle, T.; Hartmann, A.; et al. Generation and characterization of hepatocellular carcinoma cell lines with enhanced cancer stem cell potential. J. Cell Mol. Med. 2018, 22, 6238–6248. [Google Scholar] [CrossRef] [Green Version]
- Ndreshkjana, B.; Capci, A.; Klein, V.; Chanvorachote, P.; Muenzner, J.K.; Huebner, K.; Steinmann, S.; Erlenbach-Wuensch, K.; Geppert, C.I.; Agaimy, A.; et al. Combination of 5-fluorouracil and thymoquinone targets stem cell gene signature in colorectal cancer cells. Cell Death Dis. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, M.; Berral-González, A.; López-Cade, I.; Galindo-Pumariño, C.; Bueno-Fortes, S.; Martín-Merino, M.; Carrato, A.; Ocaña, A.; De La Pinta, C.; López-Alfonso, A.; et al. Cancer-associated fibroblast-derived gene signatures determine prognosis in colon cancer patients. Mol. Cancer 2021, 20, 73. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Li, H. Penalized Cox regression analysis in the high-dimensional and low-sample size settings, with applications to microarray gene expression data. Bioinformatics 2005, 21, 3001–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | RefSeq ID | Fold Change | Linear Regression Statistic | Adjusted p-Value ** | Gene Description |
|---|---|---|---|---|---|
| Upregulated genes | |||||
| VIM | NM_003380.2 | 7.85 | 1642.29 | <10 × 10−15 | Vimentin; type III intermediate filament protein |
| SPARC | NM_003118.2 | 8.27 | 1506.56 | <10 × 10−15 | Secreted prot. acidic and Cys rich; ECM component |
| NRP1 | NM_003873.5 | 3.34 | 409.97 | <10 × 10−15 | Neuropilin1; receptor prot. involved angiogenesis |
| SNAI2 | NM_003068.3 | 27.30 | 333.54 | <10 × 10−15 | Snail family transcription repressor 2; zinc finger |
| THBS1 | NM_003246.2 | 2.23 | 281.49 | <10 × 10−15 | Thrombospondin; cell-cell & cell-matrix interaction |
| TNC | NM_002160.3 | 32.20 | 251.40 | <10 × 10−15 | Tenascin C |
| PLEKHO1 | NM_016274.4 | 1.48 | 140.07 | <10 × 10−15 | Pleckstrin homology domain containing O1 |
| FGF2 | NM_002006.4 | 1.59 | 124.22 | <10 × 10−15 | Fibroblast growth factor 2 |
| FERMT2 | NM_001135000.1 | 1.33 | 113.17 | <10 × 10−15 | Fermitin family member 2 |
| SPHK2 | NM_020126.3 | 1.73 | 103.01 | <10 × 10−15 | Sphingosine kinase 2 |
| Downregulated genes | |||||
| ID2 | NM_002166.4 | −4.65 | 972.57 | <10 × 10−15 | Inhibitor DNA binding 2; negat. reg. bHLH TFs |
| TP53 | NM_000546.2 | −4.06 | 804.76 | <10 × 10−15 | Tumor protein 53; tumor suppressor |
| IL18 | NM_001562.2 | −4.79 | 734.54 | <10 × 10−15 | Interleukin 18; pro-inflammatory cytokine |
| RAMP1 | NM_005855.2 | −32.20 | 667.65 | <10 × 10−15 | Receptor activ. modif. protein 1; calcitonin GPCR |
| CDH1 | NM_004360.2 | −3.39 | 617.59 | <10 × 10−15 | Cadherin 1; cell surface adhesion molecule |
| CDKN1A * | NM_000389.2 | −3.60 | 552.82 | <10 × 10−15 | Cyclin-dependent kinase inhibitor 1A (*p21 KO) |
| ITGA3 | NM_002204.2 | −3.09 | 352.35 | <10 × 10−15 | Integrin subunit alpha 3 |
| TJP3 | NM_014428.1 | −3.25 | 350.49 | <10 × 10−15 | Tight junction protein 3 |
| S100A14 | NM_020672.1 | −3.22 | 294.47 | <10 × 10−15 | S100 calcium binding protein A14 |
| GRHL2 | NM_024915.3 | −3.89 | 291.45 | <10 × 10−15 | Grainyhead-like transcription factor 2 |
| General Function | Genes | Query Lists | Reference Lists | Enrichment p-Value | Silhouette Similarity | Functional Terms Assigned (Concurrent Enrichment) |
|---|---|---|---|---|---|---|
| Upregulated genes | ||||||
| – Fibroblast phenotype, cytoskeleton, cancer | PIK3CA, PLCG1, FGF18, FGF2, PDGFA | 5 (23) | 50 (34,208) | 1.79 × 10−10 | 0.7500 | – GO:0008543: fibroblast growth factor receptor signaling pathway (BP); Kegg: 04810: regulation of actin cytoskeleton; Kegg: 05218: melanoma. |
| – ECM changes | THBS1, SPARC, LAMC1, TNC, OLFML2B, MMP13 | 6 (23) | 134 (34,208) | 3.08 × 10−10 | 0.9129 | – GO:0031012: extracellular matrix (CC); GO:0005576: extracellular region (CC). |
| – Cell adhesion | THBS1, TNC, LAMC1, PDGFA, PIK3CA | 5 (23) | 197 (34,208) | 1.86 × 10−7 | 0.8944 | – Kegg:04510: focal adhesion. |
| Downregulated genes | ||||||
| – Cell contact regulation | CD44, ITGA2, ITGA3, ITGB4, ADAM15 PTK2B, RAMP1, DSC2 | 8 (40) | 93 (34,208) | 1.57 × 10−13 | 0.7335 | – GO:0009986: cell surface (CC); GO:0007160: cell-matrix adhesion (BP); Kegg: 04512: ECM and receptor interaction; GO:0007229: integrin-mediated signaling pathway (BP); Kegg: 05412: arrhythmogenic right ventricular cardiomyopathy. |
| – Cancer genes | CDKN1A(p21), TP53, CDH1, MET, PDGFC, TYMP | 6 (40) | 63 (34,208) | 1.12 × 10−10 | 0.7717 | – Kegg: 05218: melanoma; Kegg: 05219: bladder cancer. |
| – Cell adhesion and cytoskeleton | ITGA2, ITGA3, ITGB4, MET, PDGFC | 5 (40) | 111 (34,208) | 1.97 × 10−7 | 0.7500 | – Kegg: 04510: focal adhesion; Kegg: 04810: regulation of actin cytoskeleton. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bueno-Fortes, S.; Muenzner, J.K.; Berral-Gonzalez, A.; Hampel, C.; Lindner, P.; Berninger, A.; Huebner, K.; Kunze, P.; Bäuerle, T.; Erlenbach-Wuensch, K.; et al. A Gene Signature Derived from the Loss of CDKN1A (p21) Is Associated with CMS4 Colorectal Cancer. Cancers 2022, 14, 136. https://doi.org/10.3390/cancers14010136
Bueno-Fortes S, Muenzner JK, Berral-Gonzalez A, Hampel C, Lindner P, Berninger A, Huebner K, Kunze P, Bäuerle T, Erlenbach-Wuensch K, et al. A Gene Signature Derived from the Loss of CDKN1A (p21) Is Associated with CMS4 Colorectal Cancer. Cancers. 2022; 14(1):136. https://doi.org/10.3390/cancers14010136
Chicago/Turabian StyleBueno-Fortes, Santiago, Julienne K. Muenzner, Alberto Berral-Gonzalez, Chuanpit Hampel, Pablo Lindner, Alexandra Berninger, Kerstin Huebner, Philipp Kunze, Tobias Bäuerle, Katharina Erlenbach-Wuensch, and et al. 2022. "A Gene Signature Derived from the Loss of CDKN1A (p21) Is Associated with CMS4 Colorectal Cancer" Cancers 14, no. 1: 136. https://doi.org/10.3390/cancers14010136