
High Plus Low Dose Radiation Strategy in Combination with TIGIT and PD1 Blockade to Promote Systemic Antitumor Responses

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Drugs
2.2. Mice
2.3. Tumor Establishment and Treatments
2.4. Tumor Processing and Flow Cytometry
2.5. Statistical Analysis
3. Results
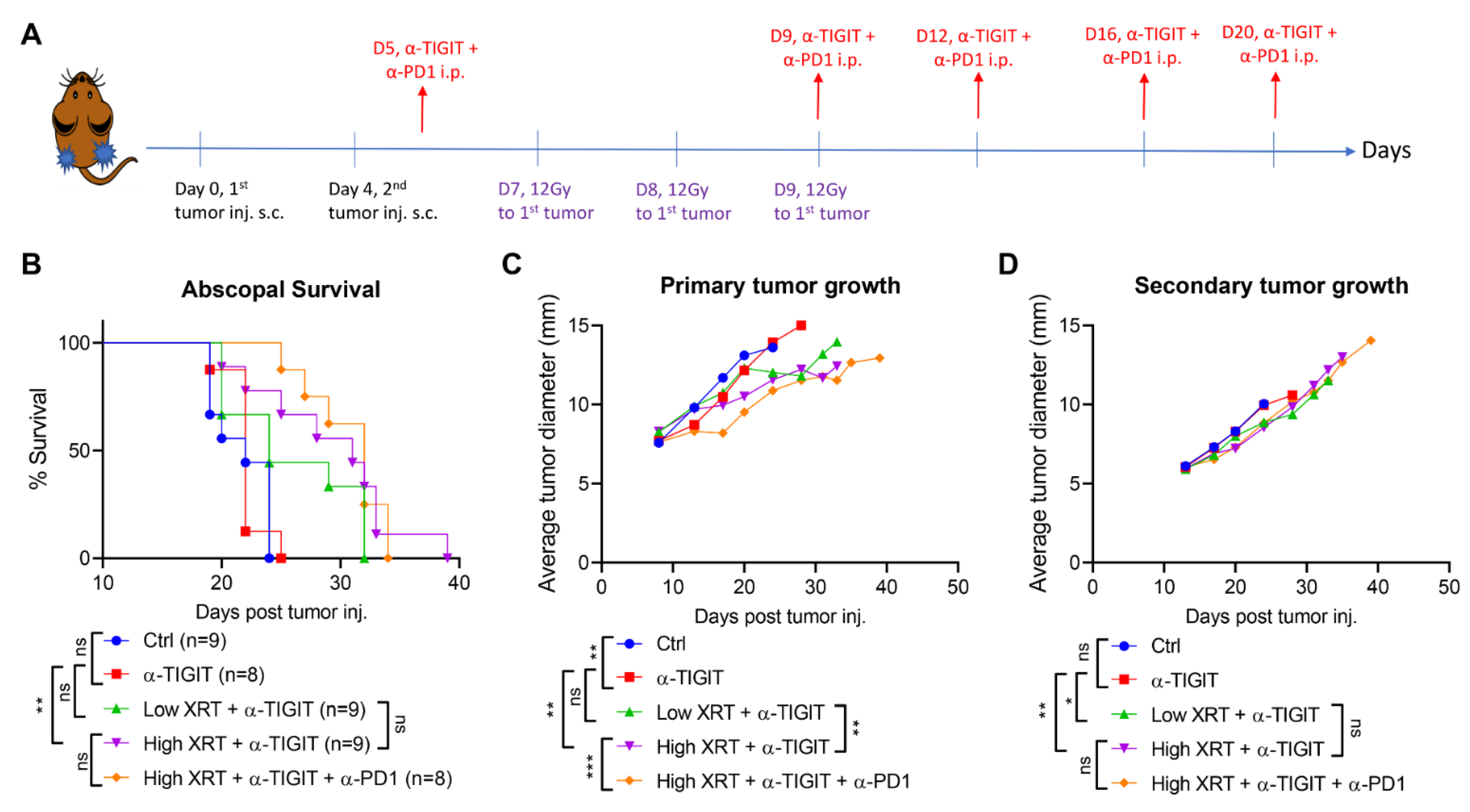
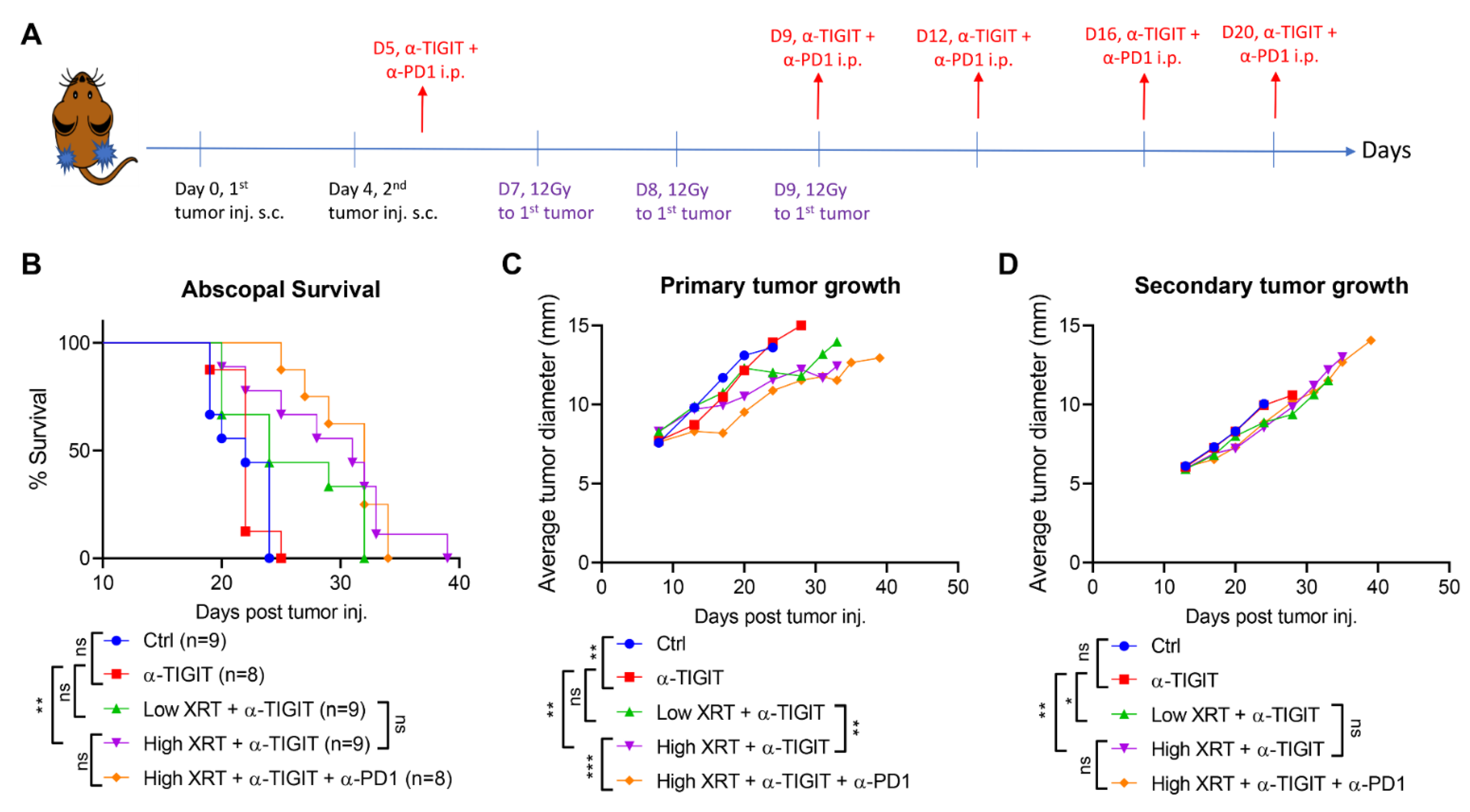
3.1. High-Dose Stereotactic Radiation with TIGIT Blockade Improves Primary and Secondary Antitumor Efficacy
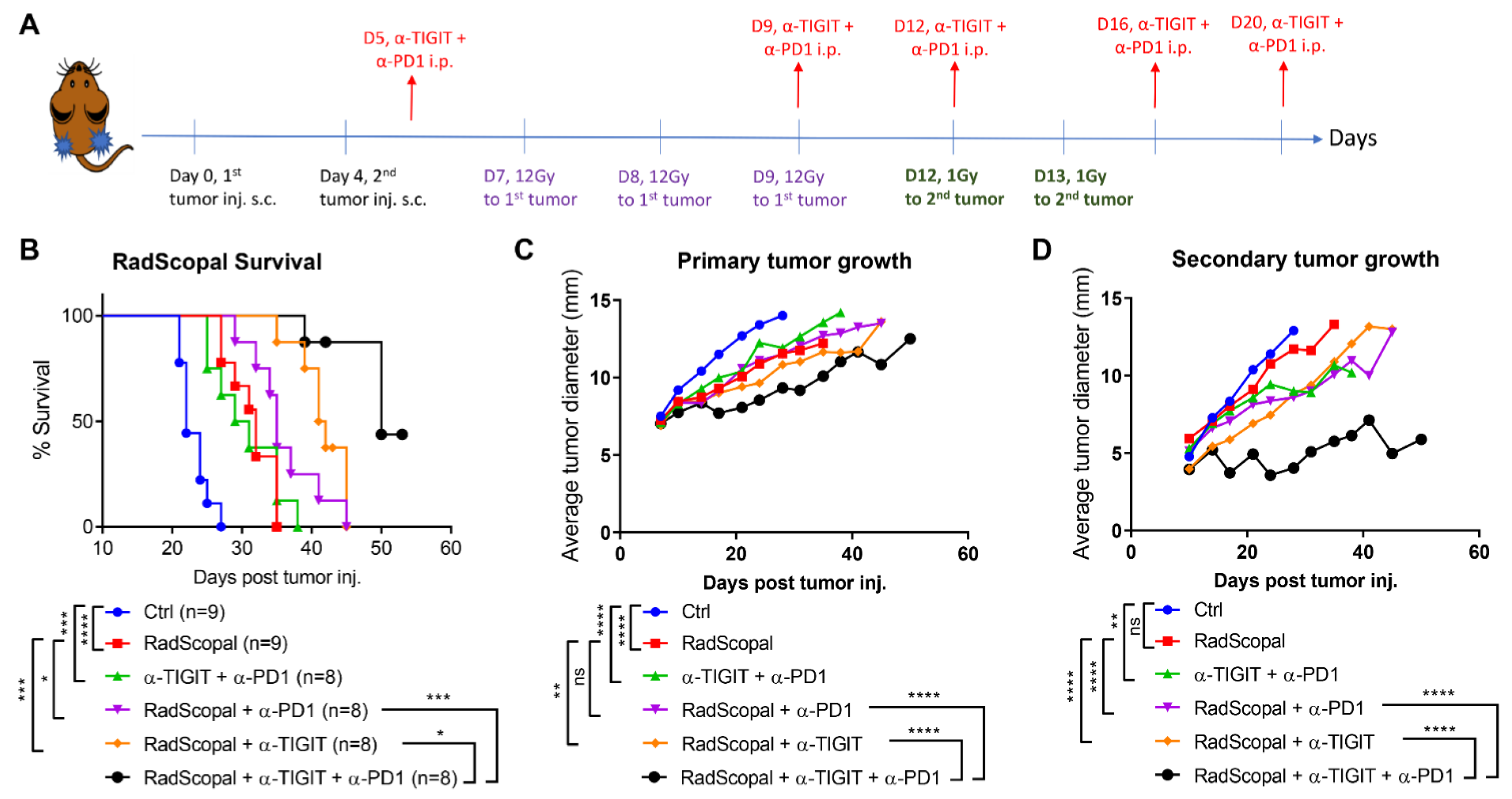
3.2. RadScopal Approach with Anti-TIGIT Plus Anti-PD1 Immunotherapy Had a High Impact on Secondary Tumors
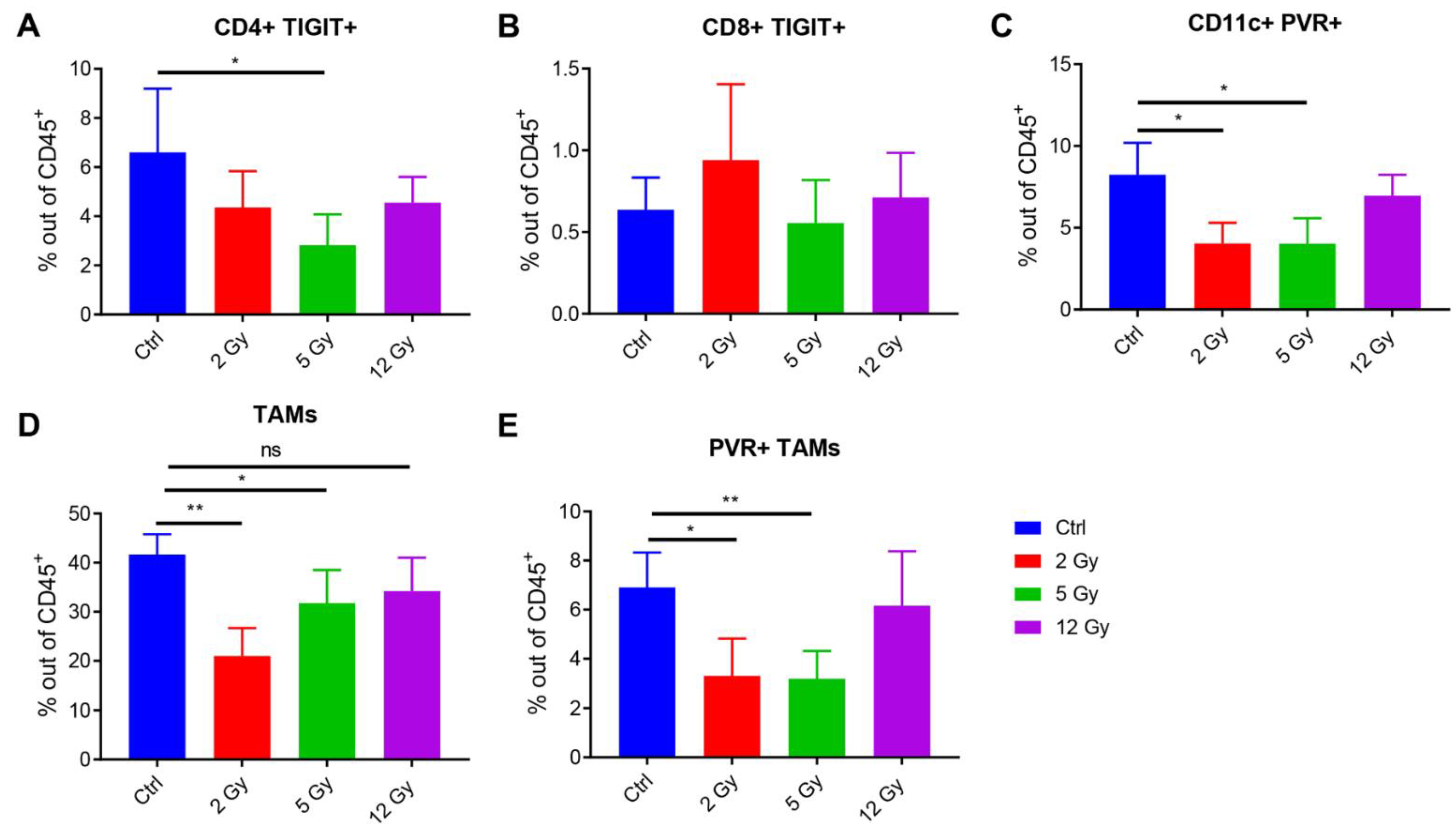
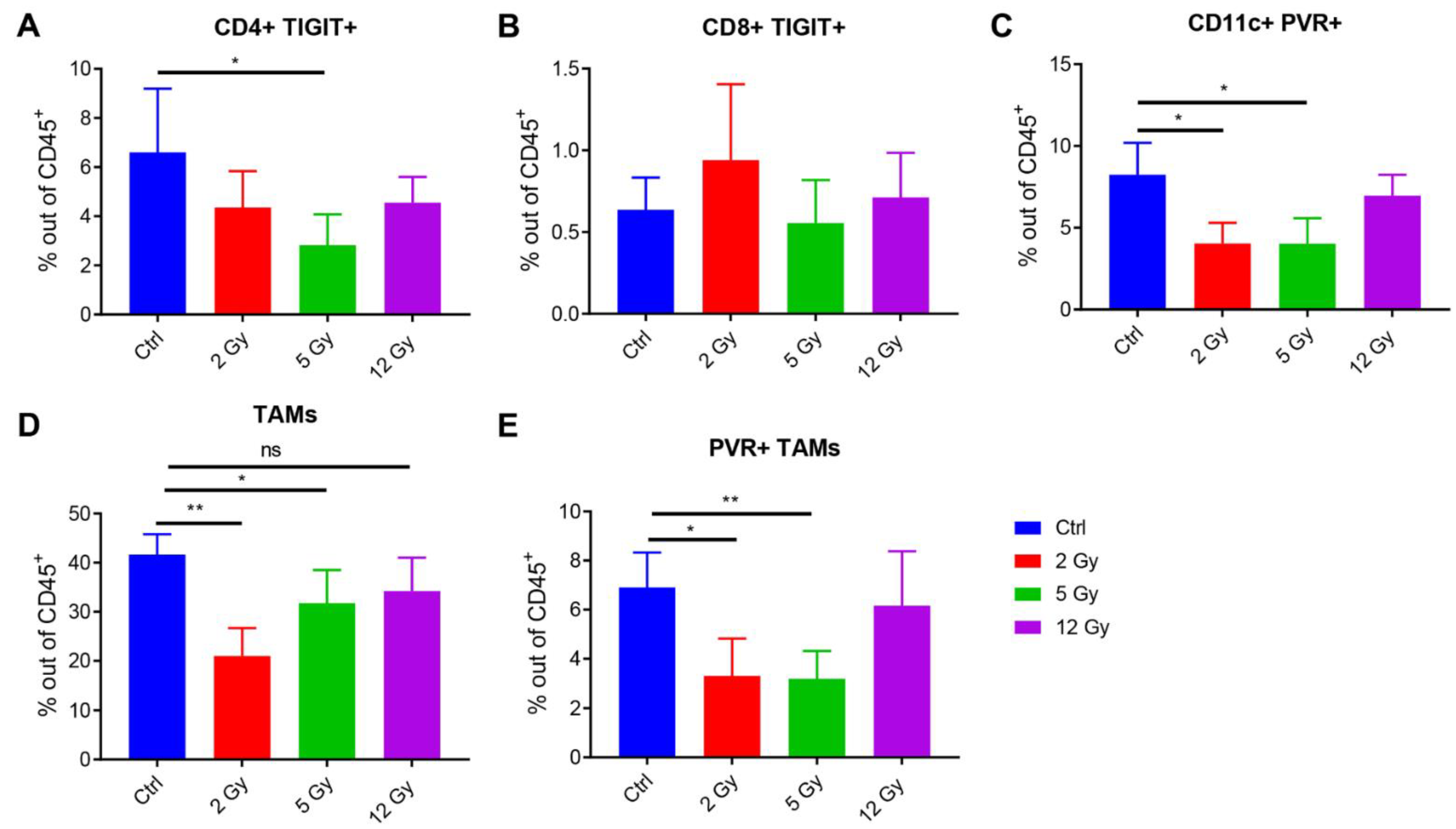
3.3. Low-Dose Radiation Reduces the TIGIT Receptor’s Expression in the TME
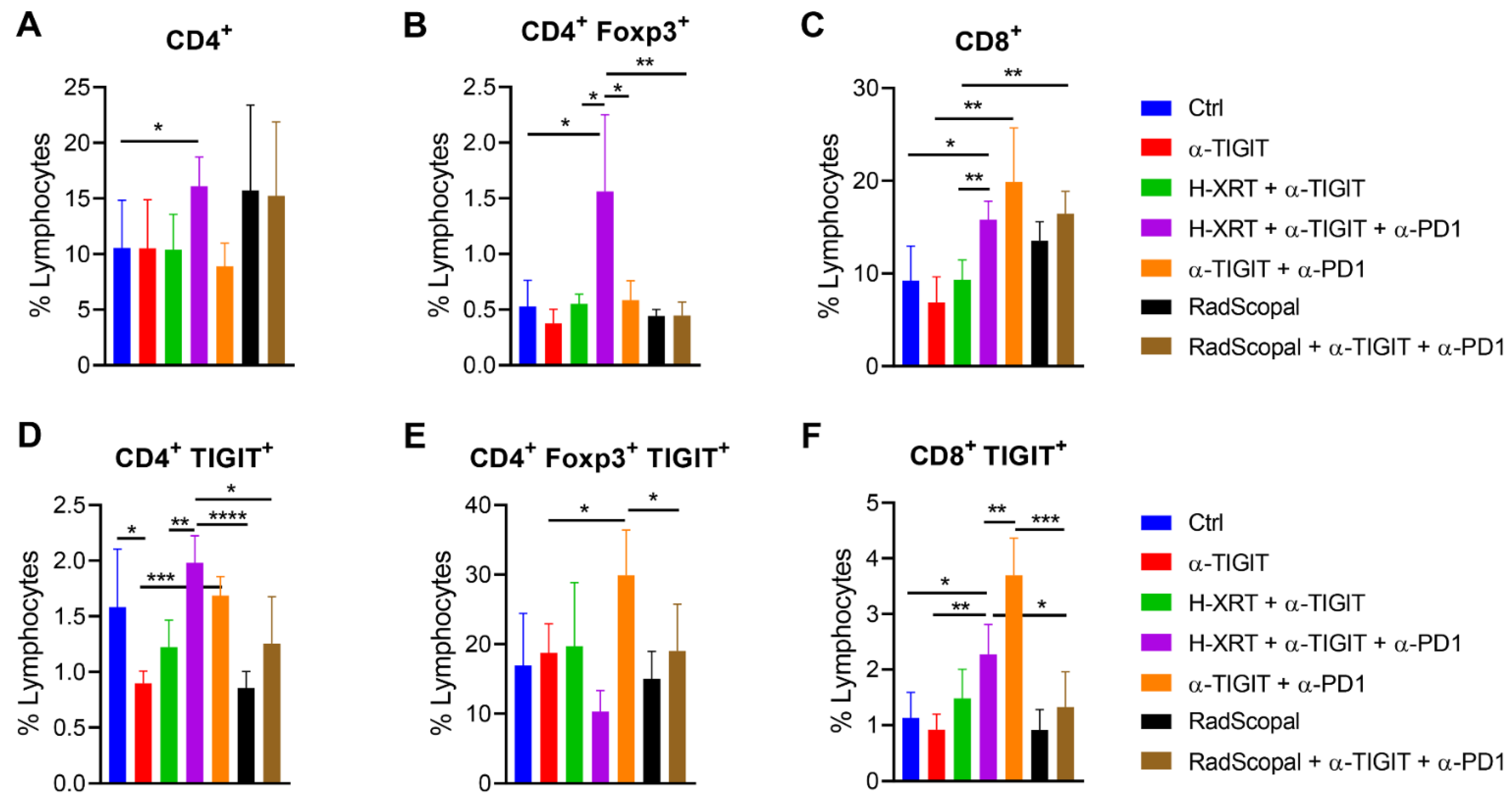
3.4. RadScopal Treatment Reduces TIGIT-Expressing T-Cell Populations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Schoenhals, J.E.; Li, A.; Valdecanas, D.R.; Ye, H.; Zang, F.; Tang, C.; Tang, M.; Liu, C.G.; Liu, X.; et al. Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy. Cancer Res. 2017, 77, 839–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niknam, S.; Barsoumian, H.B.; Schoenhals, J.E.; Jackson, H.L.; Yanamandra, N.; Caetano, M.S.; Li, A.; Younes, A.I.; Cadena, A.; Cushman, T.R.; et al. Radiation Followed by OX40 Stimulation Drives Local and Abscopal Antitumor Effects in an Anti-PD1-Resistant Lung Tumor Model. Clin. Cancer Res. 2018, 24, 5735–5743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenhals, J.E.; Cushman, T.R.; Barsoumian, H.B.; Li, A.; Cadena, A.P.; Niknam, S.; Younes, A.I.; Caetano, M.D.S.; Cortez, M.A.; Welsh, J.W. Anti-glucocorticoid-induced Tumor Necrosis Factor-Related Protein (GITR) Therapy Overcomes Radiation-Induced Treg Immunosuppression and Drives Abscopal Effects. Front. Immunol. 2018, 9, 2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formenti, S.C.; Rudqvist, N.P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Rudqvist, N.P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.W.; Emerson, R.O.; Robins, H.S.; Schneck, J.; Formenti, S.C.; Demaria, S. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol. Res. 2018, 6, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsoumian, H.B.; Ramapriyan, R.; Younes, A.I.; Caetano, M.S.; Menon, H.; Comeaux, N.I.; Cushman, T.R.; Schoenhals, J.E.; Cadena, A.P.; Reilly, T.P.; et al. Low-dose radiation treatment enhances systemic antitumor immune responses by overcoming the inhibitory stroma. J. Immunother. Cancer 2020, 8, e000537. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.R.; He, K.; Barsoumian, H.B.; Chang, J.Y.; Tang, C.; Verma, V.; Comeaux, N.; Chun, S.G.; Gandhi, S.; Truong, M.T.; et al. High-dose irradiation in combination with non-ablative low-dose radiation to treat metastatic disease after progression on immunotherapy: Results of a phase II trial. Radiother. Oncol. 2021, 162, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Rovis, T.L.; Glasner, A.; Seidel, E.; Tsukerman, P.; Yamin, R.; Enk, J.; Jonjic, S.; Mandelboim, O. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur. J. Immunol. 2013, 43, 2138–2150. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef] [PubMed]
- Harjunpaa, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtulus, S.; Sakuishi, K.; Ngiow, S.F.; Joller, N.; Tan, D.J.; Teng, M.W.; Smyth, M.J.; Kuchroo, V.K.; Anderson, A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Investig. 2015, 125, 4053–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefsson, S.E.; Beiske, K.; Blaker, Y.N.; Forsund, M.S.; Holte, H.; Ostenstad, B.; Kimby, E.; Koksal, H.; Walchli, S.; Bai, B.; et al. TIGIT and PD-1 Mark Intratumoral T Cells with Reduced Effector Function in B-cell Non-Hodgkin Lymphoma. Cancer Immunol. Res. 2019, 7, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grapin, M.; Richard, C.; Limagne, E.; Boidot, R.; Morgand, V.; Bertaut, A.; Derangere, V.; Laurent, P.A.; Thibaudin, M.; Fumet, J.D.; et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: A promising new combination. J. Immunother. Cancer 2019, 7, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.M.; Li, W.Q.; Wu, Y.H.; Han, L.; Cao, X.G.; Yang, X.M.; Wang, H.F.; Zhao, W.S.; Zhai, W.J.; Qi, Y.M.; et al. Intrinsic Expression of Immune Checkpoint Molecule TIGIT Could Help Tumor Growth in vivo by Suppressing the Function of NK and CD8(+) T Cells. Front. Immunol. 2018, 9, 2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaruzza, F.; Yeung, P.; Wang, M.; Brunner, A.; Scolan, E.L.; Cain, J.; Argast, G.; O’Young, G.; Liu, Y.; Cancilla, B.; et al. Abstract 599: Pharmacodynamic biomarkers for anti-TIGIT treatment and prevalence of TIGIT expression in multiple solid tumor types. J. Cancer Res. 2017, 77, 599. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Zhang, H.; Han, F.; Chen, X.; Lin, R.; Wang, W.; Qiu, H.; Zhuang, Z.; Liao, Q.; Zhang, W.; et al. CD155T/TIGIT Signaling Regulates CD8(+) T-cell Metabolism and Promotes Tumor Progression in Human Gastric Cancer. Cancer Res. 2017, 77, 6375–6388. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barsoumian, H.B.; Sezen, D.; Menon, H.; Younes, A.I.; Hu, Y.; He, K.; Puebla-Osorio, N.; Wasley, M.; Hsu, E.; Patel, R.R.; et al. High Plus Low Dose Radiation Strategy in Combination with TIGIT and PD1 Blockade to Promote Systemic Antitumor Responses. Cancers 2022, 14, 221. https://doi.org/10.3390/cancers14010221
Barsoumian HB, Sezen D, Menon H, Younes AI, Hu Y, He K, Puebla-Osorio N, Wasley M, Hsu E, Patel RR, et al. High Plus Low Dose Radiation Strategy in Combination with TIGIT and PD1 Blockade to Promote Systemic Antitumor Responses. Cancers. 2022; 14(1):221. https://doi.org/10.3390/cancers14010221
Chicago/Turabian StyleBarsoumian, Hampartsoum B., Duygu Sezen, Hari Menon, Ahmed I. Younes, Yun Hu, Kewen He, Nahum Puebla-Osorio, Mark Wasley, Ethan Hsu, Roshal R. Patel, and et al. 2022. "High Plus Low Dose Radiation Strategy in Combination with TIGIT and PD1 Blockade to Promote Systemic Antitumor Responses" Cancers 14, no. 1: 221. https://doi.org/10.3390/cancers14010221