
Genomic Instability and Replicative Stress in Multiple Myeloma: The Final Curtain?


{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Genomic Instability in Cancer: We Know What It Is, We Do Not Know Where It Is Coming from
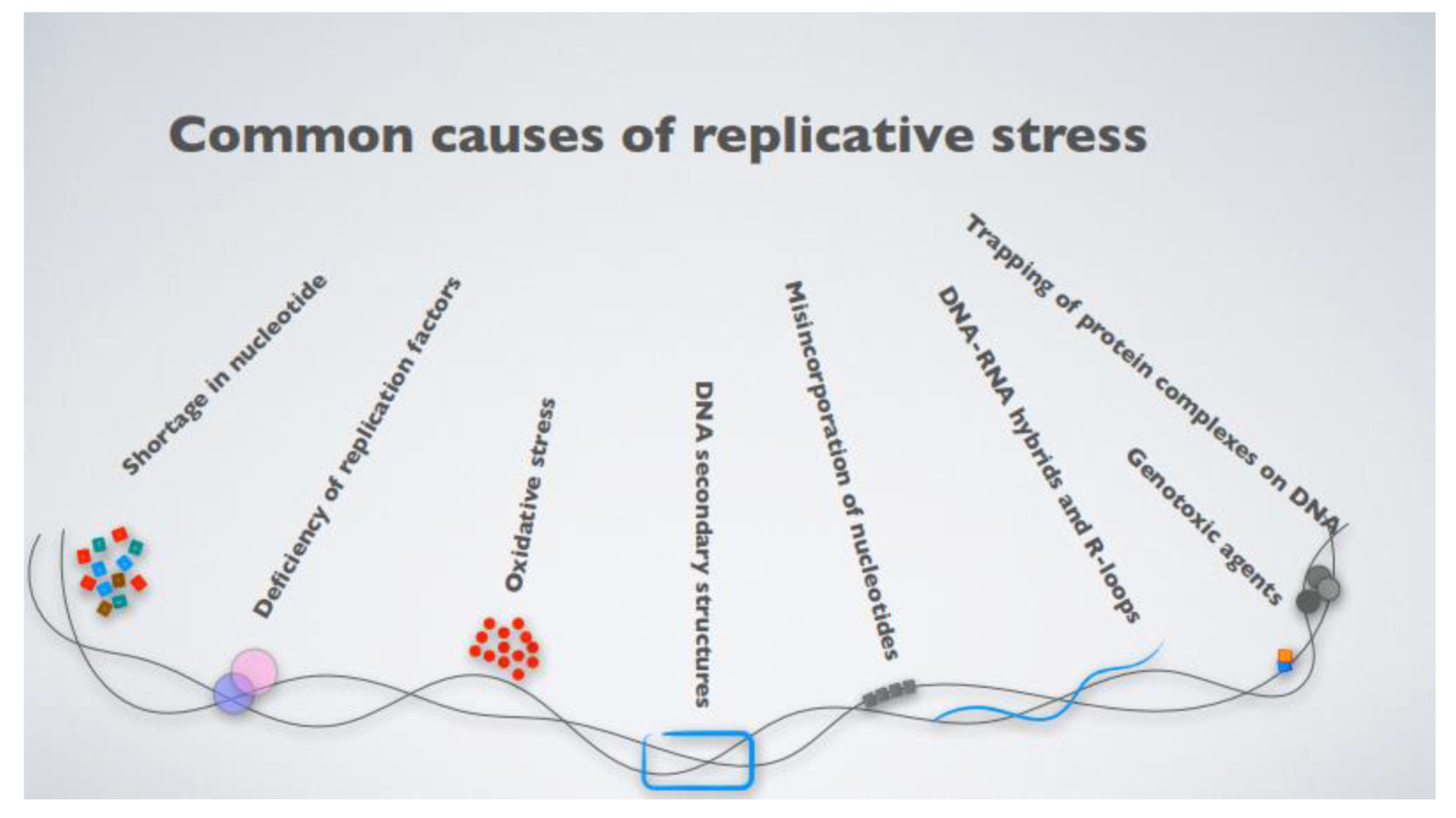
3. Replicative Stress as a Key Driver of Genomic Instability in Cancer
4. Genomic Instability and Replicative Stress in Multiple Myeloma
5. ATR, the Last Hope
6. Harnessing Replicative Stress as a Cancer Vulnerability in MM
7. Unexpected Travel Companions
8. Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.-V.; Gay, F.; Anderson, K.C. Multiple Myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Antonio, P.; Kenneth, A. Multiple Myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar]
- Pawlyn, C.; Davies, F.E. Toward Personalized Treatment in Multiple Myeloma Based on Molecular Characteristics. Blood 2019, 133, 660–675. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of Cancer Therapy: Oncogene and Non-Oncogene Addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The Proteasome Load versus Capacity Balance Determines Apoptotic Sensitivity of Multiple Myeloma Cells to Proteasome Inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef]
- Aronson, L.I.; Davies, F.E. DangER: Protein OvERload. Targeting Protein Degradation to Treat Myeloma. Haematologica 2012, 97, 1119–1130. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G., Jr. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef] [PubMed]
- Minnie, S.A.; Hill, G.R. Immunotherapy of Multiple Myeloma. J. Clin. Investig. 2020, 130, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Binder, M.; Nandakumar, B.; Rajkumar, S.V.; Kapoor, P.; Buadi, F.K.; Dingli, D.; Lacy, M.Q.; Gertz, M.A.; Hayman, S.R.; Leung, N.; et al. Mortality Trends in Multiple Myeloma after the Introduction of Novel Therapies in the United States. Leukemia 2021. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability—An Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Gao, R.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.-C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated Copy Number Evolution and Clonal Stasis in Triple-Negative Breast Cancer. Nat. Genet. 2016, 48, 1119–1130. [Google Scholar] [CrossRef]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang Model of Human Colorectal Tumor Growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated Evolution of Prostate Cancer Genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic Instability in Colorectal Cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.J.; Yang, L.-X. Gamma-H2AX—A Novel Biomarker for DNA Double-Strand Breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. ΓH2AX and Cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA Damage Checkpoint and Genomic Instability in Human Precancerous Lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-Induced Senescence Is Part of the Tumorigenesis Barrier Imposed by DNA Damage Checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Giovanni Nuciforo, P.; Bensimon, A.; et al. Oncogene-Induced Senescence Is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward Maintaining the Genome: DNA Damage and Replication Checkpoints. Annu. Rev. Genet. 2002, 36, 617–656. [Google Scholar] [CrossRef] [Green Version]
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Sulli, G.; Di Micco, R.; di Fagagna, F.A. Crosstalk between Chromatin State and DNA Damage Response in Cellular Senescence and Cancer. Nat. Rev. Cancer 2012, 12, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Lai, M.S.; Foiani, M. Preventing Replication Stress to Maintain Genome Stability: Resolving Conflicts between Replication and Transcription. Mol. Cell 2012, 45, 710–718. [Google Scholar] [CrossRef] [Green Version]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; Tornaletti, S.; D’Souza, A.D.; Hanawalt, P.C. R-Loop Generation during Transcription: Formation, Processing and Cellular Outcomes. DNA Repair 2018, 71, 69–81. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of Nucleotide Metabolism Underlies the Establishment and Maintenance of Oncogene-Induced Senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef] [Green Version]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased Global Transcription Activity as a Mechanism of Replication Stress in Cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-Transcriptional Control of DNA Replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Sankar, N.; Kadeppagari, R.-K.; Thimmapaya, B. C-Myc-Induced Aberrant DNA Synthesis and Activation of DNA Damage Response in P300 Knockdown Cells. J. Biol. Chem. 2009, 284, 15193–15205. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.V.; Dominguez-Sola, D.; Wang, L.C.; Hyrien, O.; Gautier, J. Cdc45 Is a Critical Effector of Myc-Dependent DNA Replication Stress. Cell Rep. 2013, 3, 1629–1639. [Google Scholar] [CrossRef] [Green Version]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras Oncogenes Engage Different Energy Metabolism Programs and Evoke Distinct Patterns of Oxidative and DNA Replication Stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef] [Green Version]
- Bretones, G.; Delgado, M.D.; León, J. Myc and Cell Cycle Control. Biochim. Et Biophys. Acta (BBA)—Gene Regul. Mech. 2015, 1849, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Curti, L.; Campaner, S. MYC-Induced Replicative Stress: A Double-Edged Sword for Cancer Development and Treatment. Int. J. Mol. Sci. 2021, 22, 6168. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive Oxygen Species in Cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. C-Myc Can Induce DNA Damage, Increase Reactive Oxygen Species, and Mitigate P53 Function: A Mechanism for Oncogene-Induced Genetic Instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras Proteins Induce Senescence by Altering the Intracellular Levels of Reactive Oxygen Species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [Green Version]
- Dobbelstein, M.; Sørensen, C.S. Exploiting Replicative Stress to Treat Cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Cottini, F.; Hideshima, T.; Xu, C.; Sattler, M.; Dori, M.; Agnelli, L.; ten Hacken, E.; Bertilaccio, M.T.; Antonini, E.; Neri, A.; et al. Rescue of Hippo Coactivator YAP1 Triggers DNA Damage–Induced Apoptosis in Hematological Cancers. Nat. Med. 2014, 20, 599–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottini, F.; Hideshima, T.; Suzuki, R.; Tai, Y.-T.; Bianchini, G.; Richardson, P.G.; Anderson, K.C.; Tonon, G. Synthetic Lethal Approaches Exploiting DNA Damage in Aggressive Myeloma. Cancer Discov. 2015, 5, 972–987. [Google Scholar] [CrossRef] [Green Version]
- Herrero, A.B.; Gutiérrez, N.C. Targeting Ongoing DNA Damage in Multiple Myeloma: Effects of DNA Damage Response Inhibitors on Plasma Cell Survival. Front. Oncol. 2017, 7, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero, A.B.; San Miguel, J.; Gutierrez, N.C. Deregulation of DNA Double-Strand Break Repair in Multiple Myeloma: Implications for Genome Stability. PLoS ONE 2015, 10, e0121581. [Google Scholar] [CrossRef] [PubMed]
- Botrugno, O.A.; Bianchessi, S.; Zambroni, D.; Frenquelli, M.; Belloni, D.; Bongiovanni, L.; Girlanda, S.; Di Terlizzi, S.; Ferrarini, M.; Ferrero, E.; et al. ATR Addiction in Multiple Myeloma: Synthetic Lethal Approaches Exploiting Established Therapies. Haematologica 2020, 105, 2440–2447. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.K.; Wu, X.; Tschumper, R.C.; Arendt, B.K.; Huddleston, P.M.; Henderson, K.J.; Dispenzieri, A.; Jelinek, D.F. Evidence for Ongoing DNA Damage in Multiple Myeloma Cells as Revealed by Constitutive Phosphorylation of H2AX. Leukemia 2011, 25, 1344–1353. [Google Scholar] [CrossRef]
- Dib, A.; Gabrea, A.; Glebov, O.K.; Bergsagel, P.L.; Kuehl, W.M. Characterization of MYC Translocations in Multiple Myeloma Cell Lines. J. Natl. Cancer Inst. Monogr. 2008, 2008, 25–31. [Google Scholar] [CrossRef]
- Shou, Y.; Martelli, M.L.; Gabrea, A.; Qi, Y.; Brents, L.A.; Roschke, A.; Dewald, G.; Kirsch, I.R.; Bergsagel, P.L.; Kuehl, W.M. Diverse Karyotypic Abnormalities of the C-Myc Locus Associated with c-Myc Dysregulation and Tumor Progression in Multiple Myeloma. Proc. Natl. Acad. Sci. USA 2000, 97, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Chng, W.-J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and Biological Implications of MYC Activation: A Common Difference between MGUS and Newly Diagnosed Multiple Myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affer, M.; Chesi, M.; Chen, W.D.; Keats, J.J.; Demchenko, Y.N.; Tamizhmani, K.; Garbitt, V.M.; Riggs, D.L.; Brents, L.A.; Roschke, A.V.; et al. Promiscuous MYC Locus Rearrangements Hijack Enhancers but Mostly Super-Enhancers to Dysregulate MYC Expression in Multiple Myeloma. Leukemia 2014, 28, 1725–1735. [Google Scholar] [CrossRef]
- Kuehl, W.M.; Bergsagel, P.L. MYC Addiction: A Potential Therapeutic Target in MM. Blood 2012, 120, 2351–2352. [Google Scholar] [CrossRef] [Green Version]
- Misund, K.; Keane, N.; Stein, C.K.; Asmann, Y.W.; Day, G.; Welsh, S.; Van Wier, S.A.; Riggs, D.L.; Ahmann, G.; Chesi, M.; et al. MYC Dysregulation in the Progression of Multiple Myeloma. Leukemia 2020, 34, 322–326. [Google Scholar] [CrossRef]
- Holien, T.; Våtsveen, T.K.; Hella, H.; Waage, A.; Sundan, A. Addiction to C-MYC in Multiple Myeloma. Blood 2012, 120, 2450–2453. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic Complexity of Multiple Myeloma and Its Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Liu, Y.; Wu, X.; Shell, S.M. Functions of Human Replication Protein A (RPA): From DNA Replication to DNA Damage and Stress Responses. J. Cell. Physiol. 2006, 208, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.M.R.; Lam, Z.; Last, J.I.; Byrd, P.J. Ataxia Telangiectasia: More Variation at Clinical and Cellular Levels: Ataxia Telangiectasia. Clin. Genet. 2015, 87, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Collis, S.J.; DeWeese, T.L.; Jeggo, P.A.; Parker, A.R. The Life and Death of DNA-PK. Oncogene 2005, 24, 949–961. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, M.; Ruiz-Perez, V.L.; Woods, C.G.; Jeggo, P.A.; Goodship, J.A. A Splicing Mutation Affecting Expression of Ataxia–Telangiectasia and Rad3–Related Protein (ATR) Results in Seckel Syndrome. Nat. Genet. 2003, 33, 497–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alderton, G.K.; Joenje, H.; Varon, R.; Børglum, A.D.; Jeggo, P.A.; O’Driscoll, M. Seckel Syndrome Exhibits Cellular Features Demonstrating Defects in the ATR-Signalling Pathway. Hum. Mol. Genet. 2004, 13, 3127–3138. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Baltimore, D. ATR Disruption Leads to Chromosomal Fragmentation and Early Embryonic Lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar] [CrossRef]
- De Klein, A.; Muijtjens, M.; van Os, R.; Verhoeven, Y.; Smit, B.; Carr, A.M.; Lehmann, A.R.; Hoeijmakers, J.H.J. Targeted Disruption of the Cell-Cycle Checkpoint Gene ATR Leads to Early Embryonic Lethality in Mice. Curr. Biol. 2000, 10, 479–482. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yao, F.; Marti, T.M.; Schmid, R.A.; Peng, R.-W. Beyond DNA Repair: DNA-PKcs in Tumor Metastasis, Metabolism and Immunity. Cancers 2020, 12, 3389. [Google Scholar] [CrossRef]
- Menoyo, A.; Alazzouzi, H.; Espín, E.; Armengol, M.; Yamamoto, H.; Schwartz, S. Somatic Mutations in the DNA Damage-Response Genes ATR and CHK1 in Sporadic Stomach Tumors with Microsatellite Instability. Cancer Res. 2001, 61, 7727–7730. [Google Scholar]
- Murga, M.; Bunting, S.; Montaña, M.F.; Soria, R.; Mulero, F.; Cañamero, M.; Lee, Y.; McKinnon, P.J.; Nussenzweig, A.; Fernandez-Capetillo, O. A Mouse Model of ATR-Seckel Shows Embryonic Replicative Stress and Accelerated Aging. Nat. Genet. 2009, 41, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication Stress and Cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Neelsen, K.J.; Lopes, M. Replication Fork Reversal in Eukaryotes: From Dead End to Dynamic Response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in Cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; D’Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting Oncogene-Induced Replicative Stress for the Selective Killing of Myc-Driven Tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic Stress Sensitizes Murine Cancers to Hypomorphic Suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Aye, Y.; Li, M.; Long, M.J.C.; Weiss, R.S. Ribonucleotide Reductase and Cancer: Biological Mechanisms and Targeted Therapies. Oncogene 2015, 34, 2011–2021. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, N.; Takahashi, A.; Ono, K.; Ohnishi, T. DNA Damage Induced by Alkylating Agents and Repair Pathways. J. Nucleic Acids 2010, 2010, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR Kinase Activities by the Radiosensitizing Agent, Caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar]
- Nishida, H.; Tatewaki, N.; Nakajima, Y.; Magara, T.; Ko, K.M.; Hamamori, Y.; Konishi, T. Inhibition of ATR Protein Kinase Activity by Schisandrin B in DNA Damage Response. Nucleic Acids Res. 2009, 37, 5678–5689. [Google Scholar] [CrossRef] [Green Version]
- Toledo, L.I.; Murga, M.; Gutierrez-Martinez, P.; Soria, R.; Fernandez-Capetillo, O. ATR Signaling Can Drive Cells into Senescence in the Absence of DNA Breaks. Genes Dev. 2008, 22, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A Cell-Based Screen Identifies ATR Inhibitors with Synthetic Lethal Properties for Cancer-Associated Mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charrier, J.-D.; Durrant, S.J.; Golec, J.M.C.; Kay, D.P.; Knegtel, R.M.A.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of Potent and Selective Inhibitors of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Protein Kinase as Potential Anticancer Agents. J. Med. Chem. 2011, 54, 2320–2330. [Google Scholar] [CrossRef]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in Vivo Using the Novel Inhibitor VE-822 Results in Selective Sensitization of Pancreatic Tumors to Radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [Green Version]
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4-{4-[(3 R )-3-Methylmorpholin-4-Yl]-6-[1-(Methylsulfonyl)Cyclopropyl]Pyrimidin-2-Yl}-1 H -Indole (AZ20): A Potent and Selective Inhibitor of ATR Protein Kinase with Monotherapy in Vivo Antitumor Activity. J. Med. Chem. 2013, 56, 2125–2138. [Google Scholar] [CrossRef]
- Vendetti, F.P.; Lau, A.; Schamus, S.; Conrads, T.P.; O’Connor, M.J.; Bakkenist, C.J. The Orally Active and Bioavailable ATR Kinase Inhibitor AZD6738 Potentiates the Anti-Tumor Effects of Cisplatin to Resolve ATM-Deficient Non-Small Cell Lung Cancer In Vivo. Oncotarget 2015, 6, 44289–44305. [Google Scholar] [CrossRef] [Green Version]
- Zenke, F.T.; Zimmermann, A.; Dahmen, H.; Elenbaas, B.; Pollard, J.; Reaper, P.; Bagrodia, S.; Spilker, M.E.; Amendt, C.; Blaukat, A. Abstract 369: Antitumor Activity of M4344, a Potent and Selective ATR Inhibitor, in Monotherapy and Combination Therapy. In Experimental and Molecular Therapeutics; American Association for Cancer Research: Philadelphia, PA, USA, 2019; p. 369. [Google Scholar]
- Jo, U.; Senatorov, I.S.; Zimmermann, A.; Saha, L.K.; Murai, Y.; Kim, S.H.; Rajapakse, V.N.; Elloumi, F.; Takahashi, N.; Schultz, C.W.; et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells With Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol. Cancer Ther. 2021, 20, 1431–1441. [Google Scholar] [CrossRef]
- Lücking, U.; Wortmann, L.; Wengner, A.M.; Lefranc, J.; Lienau, P.; Briem, H.; Siemeister, G.; Bömer, U.; Denner, K.; Schäfer, M.; et al. Damage Incorporated: Discovery of the Potent, Highly Selective, Orally Available ATR Inhibitor BAY 1895344 with Favorable Pharmacokinetic Properties and Promising Efficacy in Monotherapy and in Combination Treatments in Preclinical Tumor Models. J. Med. Chem. 2020, 63, 7293–7325. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage–Inducing or Repair–Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngoi, N.Y.L.; Pham, M.M.; Tan, D.S.P.; Yap, T.A. Targeting the Replication Stress Response through Synthetic Lethal Strategies in Cancer Medicine. Trends Cancer 2021, 7, 930–957. [Google Scholar] [CrossRef]
- Thomas, A.; Redon, C.E.; Sciuto, L.; Padiernos, E.; Ji, J.; Lee, M.-J.; Yuno, A.; Lee, S.; Zhang, Y.; Tran, L.; et al. Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. JCO 2018, 36, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; de Miguel Luken, M.J.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. JCO 2020, 38, 3195–3204. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Wesolowski, R.; Devoe, C.; Lord, S.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R.; Yap, T.A. Phase 1 Study of the ATR Inhibitor Berzosertib in Combination with Cisplatin in Patients with Advanced Solid Tumours. Br. J. Cancer 2021, 125, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 Study of the ATR Inhibitor Berzosertib (Formerly M6620, VX-970) Combined with Gemcitabine ± Cisplatin in Patients with Advanced Solid Tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR Inhibitors as a Synthetic Lethal Therapy for Tumours Deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef]
- Tsai, S.; Fournier, L.-A.; Chang, E.Y.; Wells, J.P.; Minaker, S.W.; Zhu, Y.D.; Wang, A.Y.-H.; Wang, Y.; Huntsman, D.G.; Stirling, P.C. ARID1A Regulates R-Loop Associated DNA Replication Stress. PLoS Genet. 2021, 17, e1009238. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 Inhibitors and Cancer Therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Sanjiv, K.; Hagenkort, A.; Calderón-Montaño, J.M.; Koolmeister, T.; Reaper, P.M.; Mortusewicz, O.; Jacques, S.A.; Kuiper, R.V.; Schultz, N.; Scobie, M.; et al. Cancer-Specific Synthetic Lethality between ATR and CHK1 Kinase Activities. Cell Rep. 2016, 14, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Landau, H.J.; McNeely, S.C.; Nair, J.S.; Comenzo, R.L.; Asai, T.; Friedman, H.; Jhanwar, S.C.; Nimer, S.D.; Schwartz, G.K. The Checkpoint Kinase Inhibitor AZD7762 Potentiates Chemotherapy-Induced Apoptosis of P53-Mutated Multiple Myeloma Cells. Mol. Cancer Ther. 2012, 11, 1781–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kumagai, A.; Dunphy, W.G. Positive Regulation of Wee1 by Chk1 and 14-3-3 Proteins. MBoC 2001, 12, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, K.; Doroshow, J.H.; Kummar, S. Wee1 Kinase as a Target for Cancer Therapy. Cell Cycle 2013, 12, 3348–3353. [Google Scholar] [CrossRef] [Green Version]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-Dependent Kinase Suppression by WEE1 Kinase Protects the Genome through Control of Replication Initiation and Nucleotide Consumption. Mol. Cell Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, S.X.; Markkanen, E.; Jiang, Y.; Sarkar, S.; Woodcock, M.; Orlando, G.; Mavrommati, I.; Pai, C.-C.; Zalmas, L.-P.; Drobnitzky, N.; et al. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by DNTP Starvation. Cancer Cell 2015, 28, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, R.S.S.; Dantonio, P.M.; Guimarães, T.; de Oliveira, M.B.; Fook Alves, V.L.; Sandes, A.F.; Fernando, R.C.; Colleoni, G.W.B. Sequential Combination of Bortezomib and WEE1 Inhibitor, MK-1775, Induced Apoptosis in Multiple Myeloma Cell Lines. Biochem. Biophys. Res. Commun. 2019, 519, 597–604. [Google Scholar] [CrossRef]
- Liang, L.; He, Y.; Wang, H.; Zhou, H.; Xiao, L.; Ye, M.; Kuang, Y.; Luo, S.; Zuo, Y.; Feng, P.; et al. The Wee1 Kinase Inhibitor MK1775 Suppresses Cell Growth, Attenuates Stemness and Synergises with Bortezomib in Multiple Myeloma. Br. J. Haematol. 2020, 191, 62–76. [Google Scholar] [CrossRef]
- Tabayashi, T.; Tanaka, Y.; Takahashi, Y.; Kimura, Y.; Tomikawa, T.; Sagawa, M.; Anan-Nemoto, T.; Watanabe, R.; Tokuhira, M.; Kizaki, M. Inhibition of WEE1 Induces Cell Death of Both Bortezomib- and Lenalidomide-Resistant Multiple Myeloma Cells: A Novel Therapeutic Approach Targeting Cell-Cycle Checkpoint Kinase. Blood 2016, 128, 3256. [Google Scholar] [CrossRef]
- Xing, L.; Lin, L.; Yu, T.; Li, Y.; Cho, S.-F.; Liu, J.; Wen, K.; Hsieh, P.A.; Kinneer, K.; Munshi, N.; et al. A Novel BCMA PBD-ADC with ATM/ATR/WEE1 Inhibitors or Bortezomib Induce Synergistic Lethality in Multiple Myeloma. Leukemia 2020, 34, 2150–2162. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Min, W.; Bruhn, C.; Grigaravicius, P.; Zhou, Z.-W.; Li, F.; Krüger, A.; Siddeek, B.; Greulich, K.-O.; Popp, O.; Meisezahl, C.; et al. Poly(ADP-Ribose) Binding to Chk1 at Stalled Replication Forks Is Required for S-Phase Checkpoint Activation. Nat. Commun. 2013, 4, 2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to Poly(ADP-Ribose) Polymerase Inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W.G. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic Lethality as an Engine for Cancer Drug Target Discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness Revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Gourzones, C.; Bret, C.; Moreaux, J. Treatment May Be Harmful: Mechanisms/Prediction/Prevention of Drug-Induced DNA Damage and Repair in Multiple Myeloma. Front. Genet. 2019, 10, 861. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, D.A.; et al. Bortezomib-Induced “BRCAness” Sensitizes Multiple Myeloma Cells to PARP Inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagpulinsa, D.A.; Ayyadevara, S.; Yaccoby, S.; Reis, R.J.S. A Cyclin-Dependent Kinase Inhibitor, Dinaciclib, Impairs Homologous Recombination and Sensitizes Multiple Myeloma Cells to PARP Inhibition. Mol. Cancer Ther. 2016, 15, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caracciolo, D.; Scionti, F.; Juli, G.; Altomare, E.; Golino, G.; Todoerti, K.; Grillone, K.; Riillo, C.; Arbitrio, M.; Iannone, M.; et al. Exploiting MYC-Induced PARPness to Target Genomic Instability in Multiple Myeloma. Haematologica 2020, 106, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Pommier, Y. PARP Trapping Beyond Homologous Recombination and Platinum Sensitivity in Cancers. Annu. Rev. Cancer Biol. 2019, 3, 131–150. [Google Scholar] [CrossRef]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RecQ Helicases in DNA Repair, Recombination, and Replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [Green Version]
- Veith, S.; Mangerich, A. RecQ Helicases and PARP1 Team up in Maintaining Genome Integrity. Ageing Res. Rev. 2015, 23, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Viziteu, E.; Klein, B.; Basbous, J.; Lin, Y.-L.; Hirtz, C.; Gourzones, C.; Tiers, L.; Bruyer, A.; Vincent, L.; Grandmougin, C.; et al. RECQ1 Helicase Is Involved in Replication Stress Survival and Drug Resistance in Multiple Myeloma. Leukemia 2017, 31, 2104–2113. [Google Scholar] [CrossRef] [Green Version]
- Choe, K.N.; Nicolae, C.M.; Constantin, D.; Kawasawa, Y.I.; Delgado-Diaz, M.R.; De, S.; Freire, R.; Smits, V.A.J.; Moldovan, G.-L. HUWE1 Interacts with PCNA to Alleviate Replication Stress. EMBO Rep. 2016, 17, 874–886. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, K.B.; Bang, S.; Kurokawa, M.; Gerber, S.A. Direct Regulation of Chk1 Protein Stability by E3 Ubiquitin Ligase HUWE1. FEBS J. 2020, 287, 1985–1999. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Ma.avrommatis, K.; Wardell, C.P.; Cody Ashby, T.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of Novel Mutational Drivers Reveals Oncogene Dependencies in Multiple Myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Kunz, V.; Bommert, K.S.; Kruk, J.; Schwinning, D.; Chatterjee, M.; Stühmer, T.; Bargou, R.; Bommert, K. Targeting of the E3 Ubiquitin-Protein Ligase HUWE1 Impairs DNA Repair Capacity and Tumor Growth in Preclinical Multiple Myeloma Models. Sci. Rep. 2020, 10, 18419. [Google Scholar] [CrossRef]
- Shammas, M.A.; Shmookler Reis, R.J.; Koley, H.; Batchu, R.B.; Li, C.; Munshi, N.C. Dysfunctional Homologous Recombination Mediates Genomic Instability and Progression in Myeloma. Blood 2009, 113, 2290–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, M.K.; Buckanovich, R.J.; Bernstein, K.A. Regulation and Pharmacological Targeting of RAD51 in Cancer. NAR Cancer 2020, 2, zcaa024. [Google Scholar] [CrossRef] [PubMed]
- Alagpulinsa, D.A.; Yaccoby, S.; Ayyadevara, S.; Shmookler Reis, R.J. A Peptide Nucleic Acid Targeting Nuclear RAD51 Sensitizes Multiple Myeloma Cells to Melphalan Treatment. Cancer Biol. Ther. 2015, 16, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Alagpulinsa, D.A.; Ayyadevara, S.; Shmookler Reis, R.J. A Small-Molecule Inhibitor of RAD51 Reduces Homologous Recombination and Sensitizes Multiple Myeloma Cells to Doxorubicin. Front. Oncol. 2014, 4, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, L.; Krauth, M.T.; Podar, K.; Raab, M.-S. Pathway-Directed Therapy in Multiple Myeloma. Cancers 2021, 13, 1668. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botrugno, O.A.; Tonon, G. Genomic Instability and Replicative Stress in Multiple Myeloma: The Final Curtain? Cancers 2022, 14, 25. https://doi.org/10.3390/cancers14010025
Botrugno OA, Tonon G. Genomic Instability and Replicative Stress in Multiple Myeloma: The Final Curtain? Cancers. 2022; 14(1):25. https://doi.org/10.3390/cancers14010025
Chicago/Turabian StyleBotrugno, Oronza A., and Giovanni Tonon. 2022. "Genomic Instability and Replicative Stress in Multiple Myeloma: The Final Curtain?" Cancers 14, no. 1: 25. https://doi.org/10.3390/cancers14010025