
Upregulation of CD36, a Fatty Acid Translocase, Promotes Colorectal Cancer Metastasis by Increasing MMP28 and Decreasing E-Cadherin Expression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
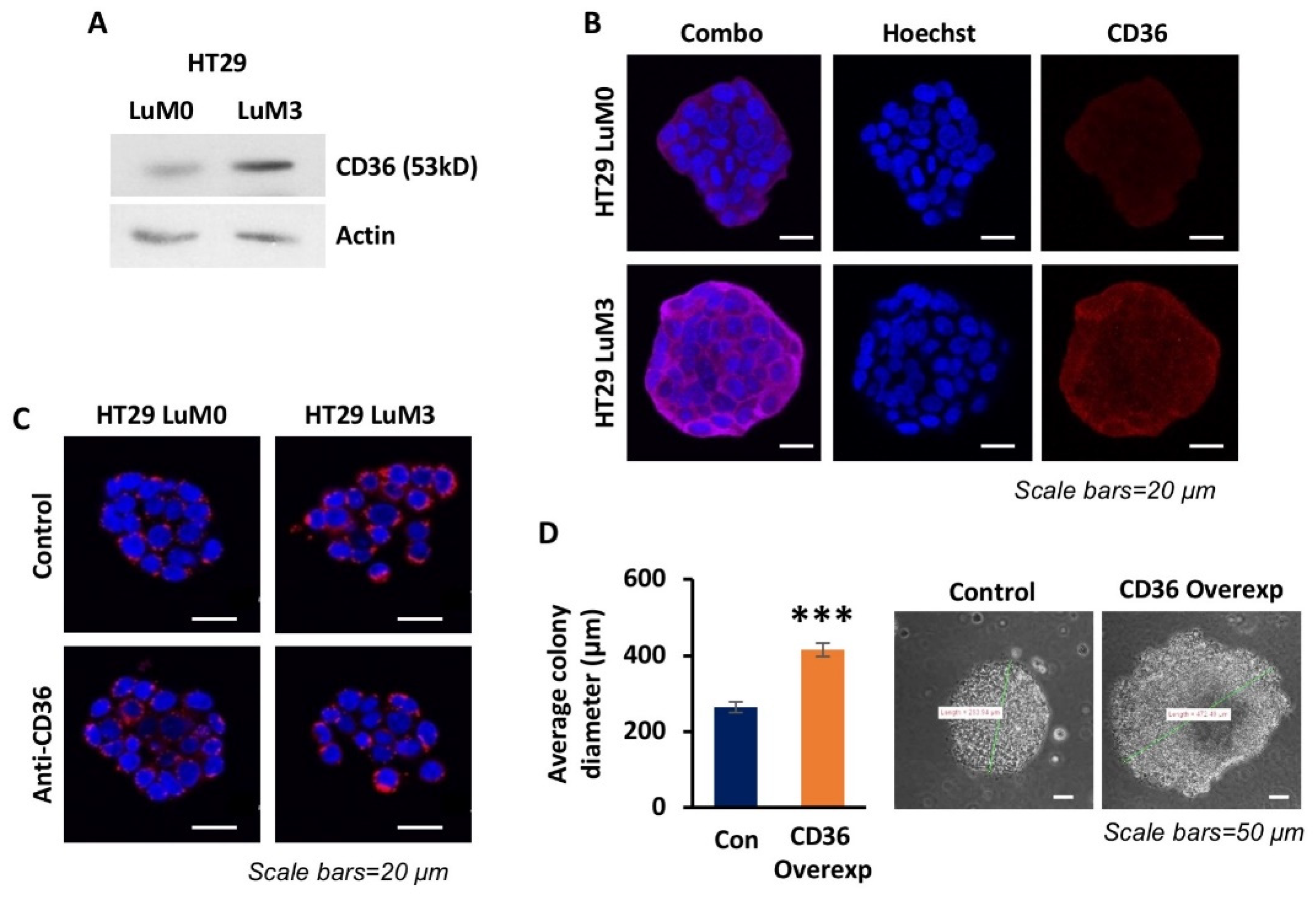
3.1. CD36 Promotes Invasion and Colony Formation in HCT116 Cells
3.2. High Expression of CD36 Is Associated with a More Metastatic Phenotype in Isogenic CRC Cell Lines
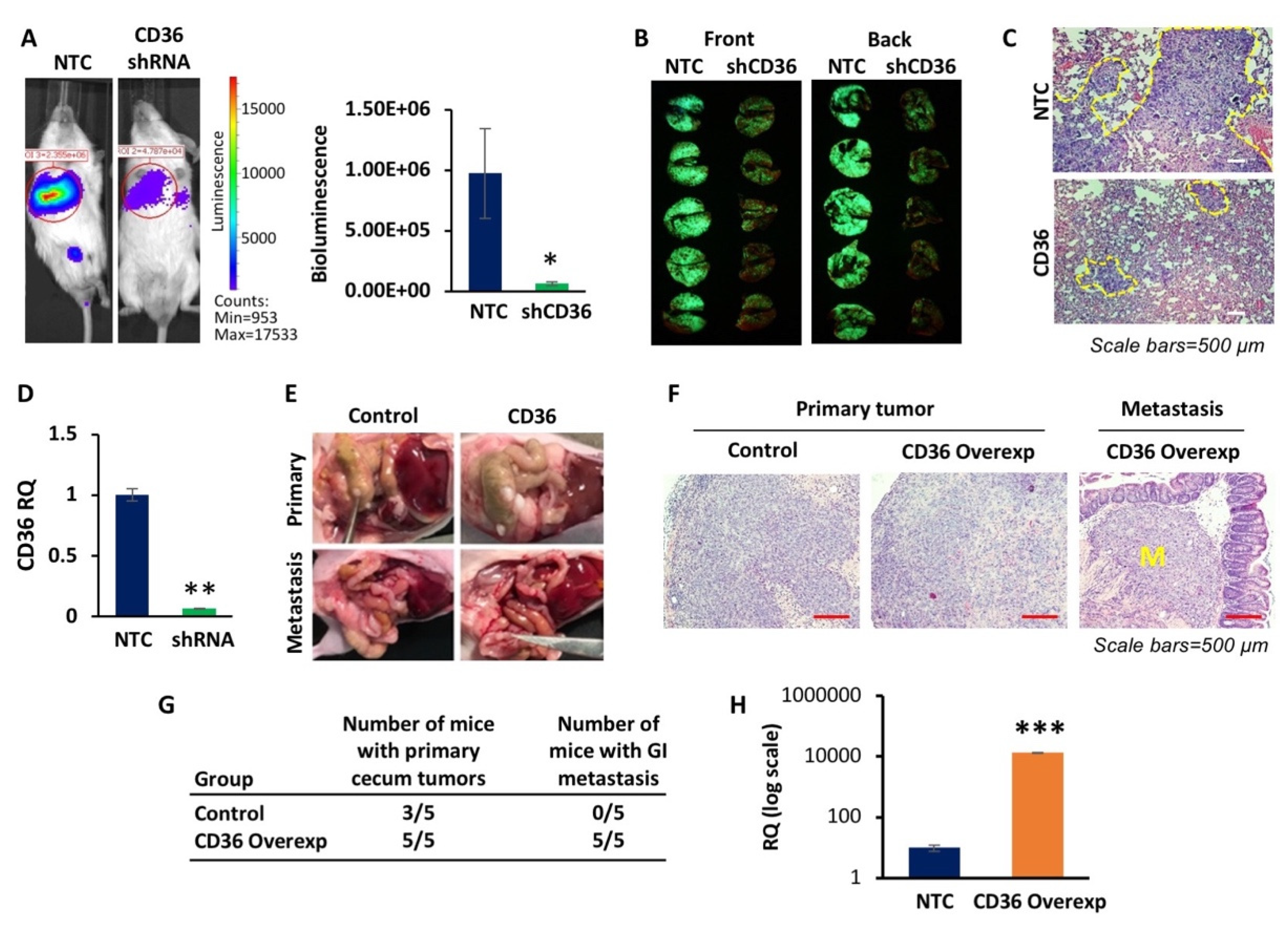
3.3. CD36 Promotes Lung Colonization and Orthotopic Metastasis In Vivo
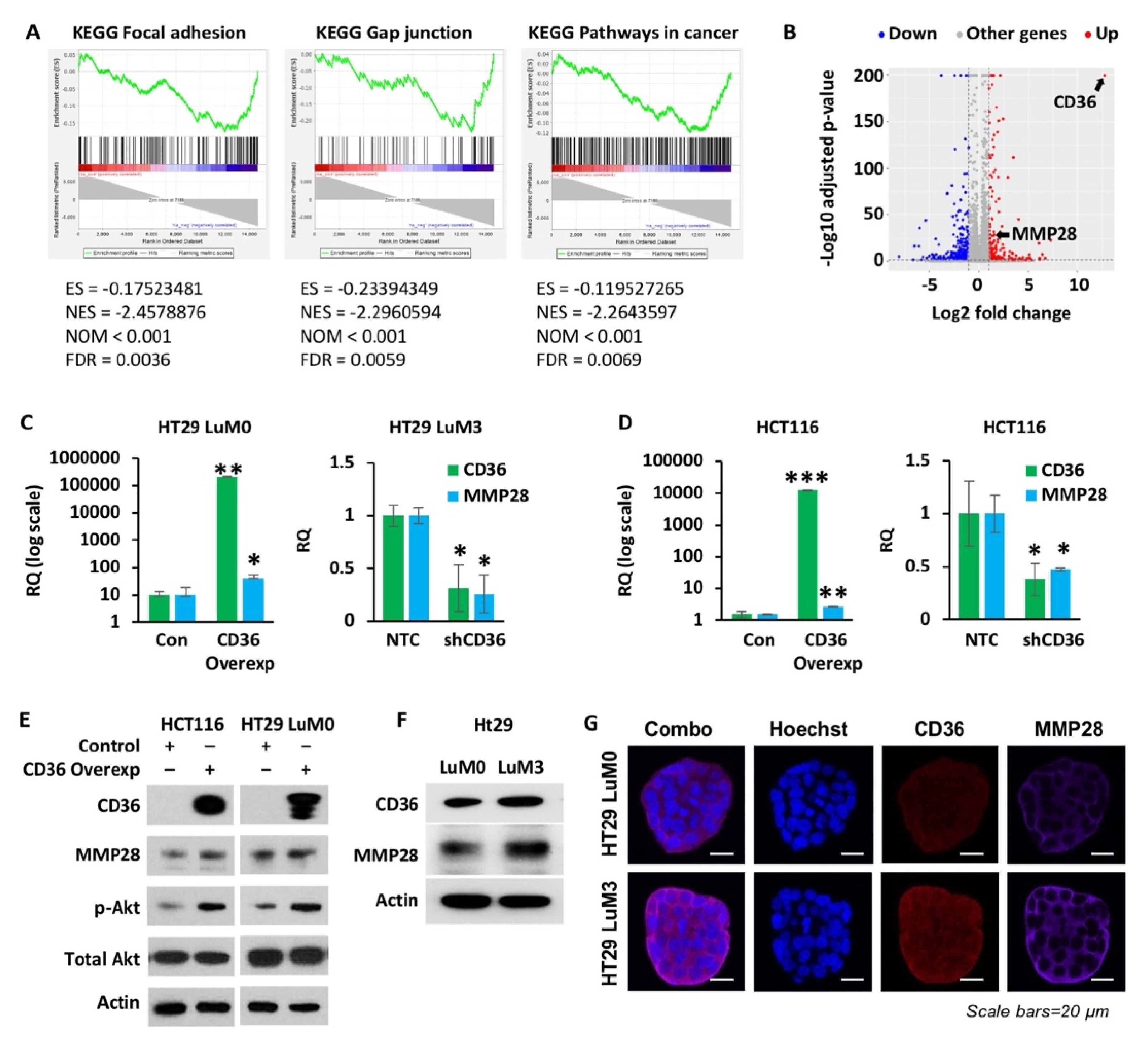
3.4. High Expression of CD36 Increases MMP28 Expression
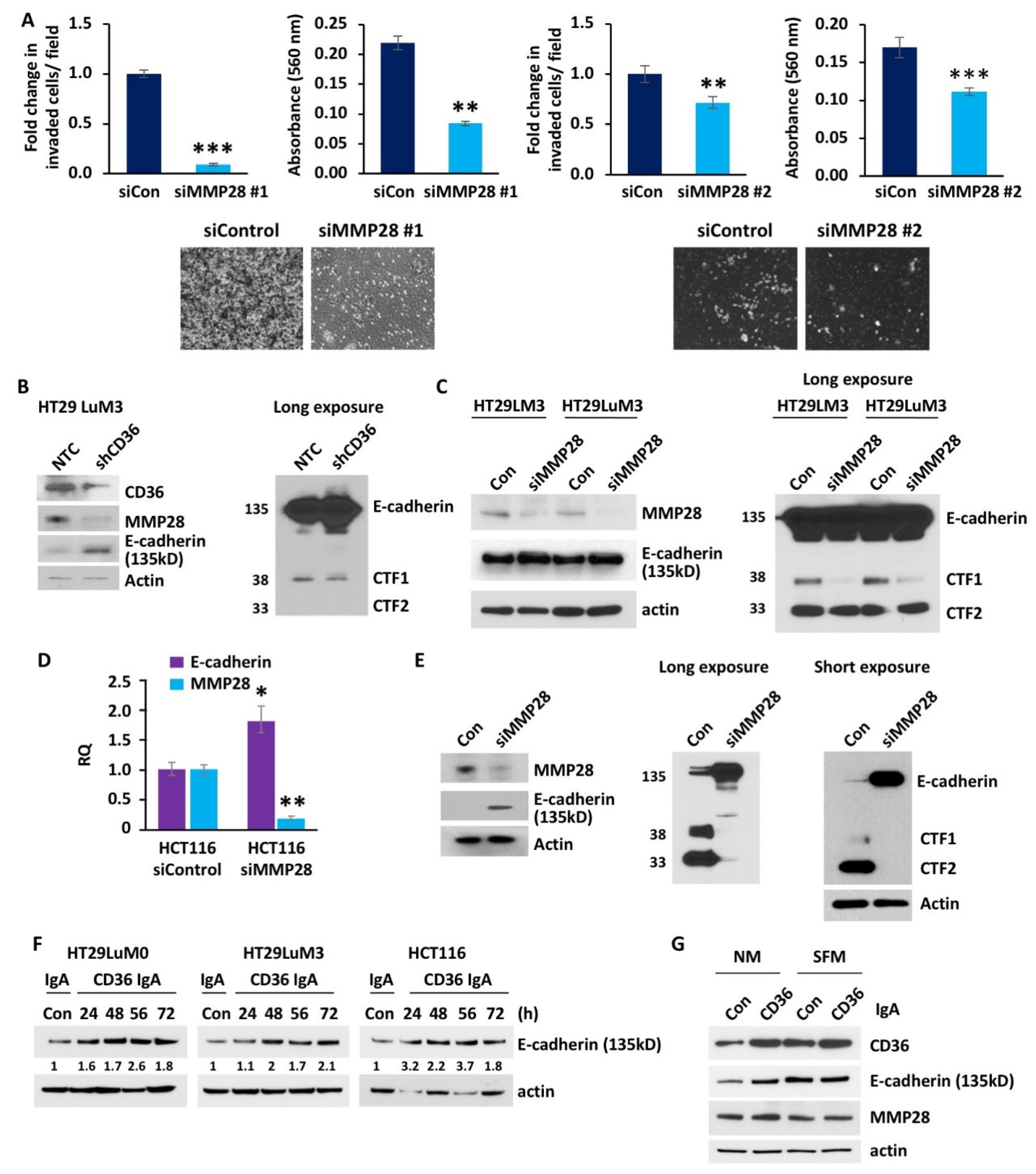
3.5. MMP28 Promotes CRC Cell Invasion and Reduces Expression of E-Cadherin In Vitro
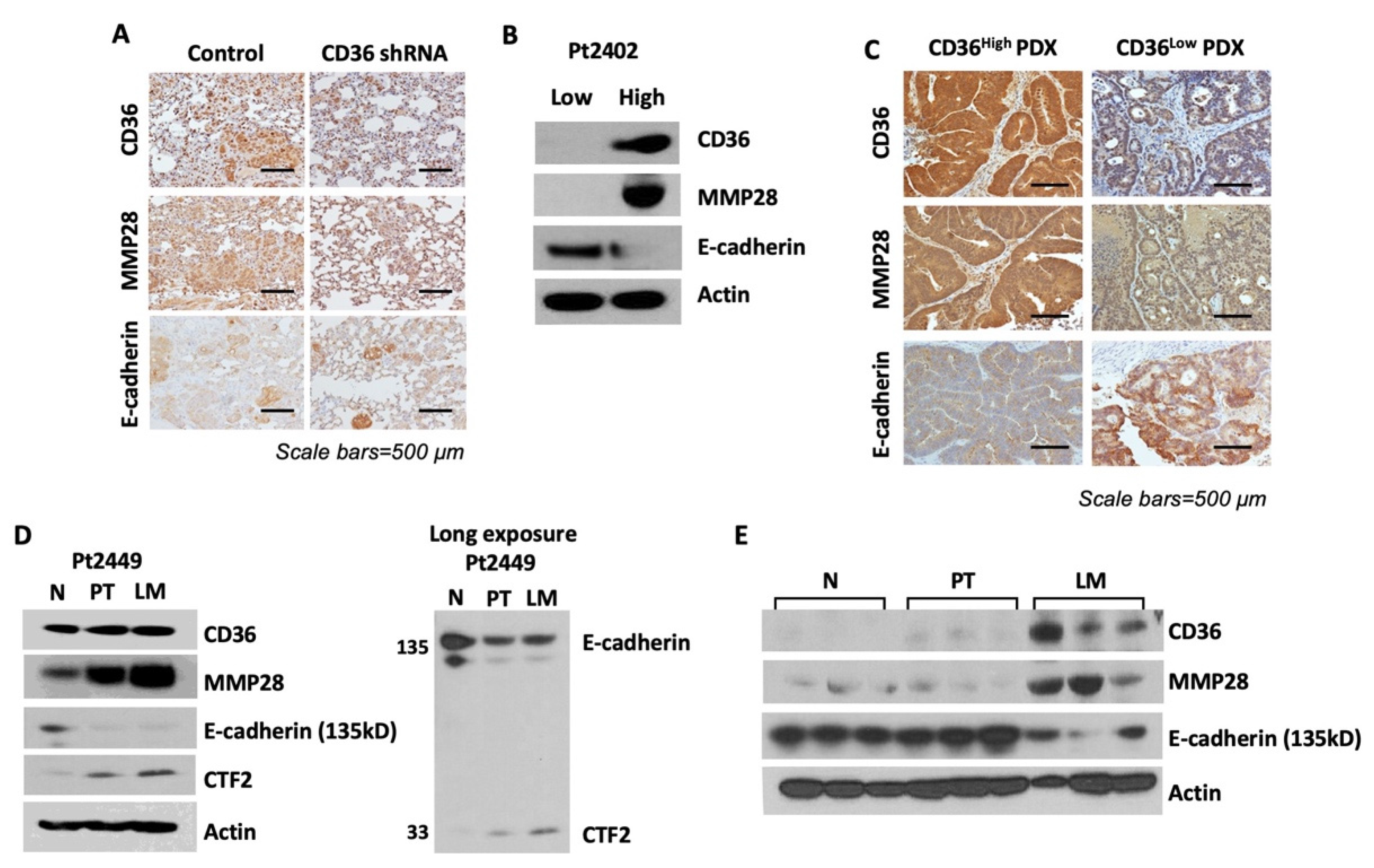
3.6. Overexpression of CD36 Is Associated with an Increase in MMP28 Expression and Reduction of E-Cadherin In Vivo and Human CRC Specimens
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Shin, H.-R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Cancer Society. Cancer Facts & Figures. 2021. Available online: https://www.cancer.org/cancer/colon-rectal-cancer/detection-diagnosis-staging/survival-rates.html#references (accessed on 15 March 2021).
- Howlader, N.A.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; Chen, H.S.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2017; National Cancer Institute: Bethesda, MD, USA, 2020. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 7 March 2021).
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, D.; Duke, G.; Heuer, T.S.; O’Farrell, M.; Wagman, A.S.; McCulloch, W.; Kemble, G. Fatty acid synthase—Modern tumor cell biology insights into a classical oncology target. Pharmacol. Ther. 2017, 177, 23–31. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-Function of CD36 and Importance of Fatty Acid Signal Transduction in Fat Metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef] [Green Version]
- Glatz, J.F.C.; Luiken, J.J.F.P. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Drover, V.A.; Nguyen, D.V.; Bastie, C.; Darlington, Y.F.; Abumrad, N.A.; Pessin, J.E.; London, E.; Sahoo, D.; Phillips, M.C. CD36 Mediates Both Cellular Uptake of Very Long Chain Fatty Acids and Their Intestinal Absorption in Mice. J. Biol. Chem. 2008, 283, 13108–13115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef]
- Jayawardhana, A.M.D.S.; Stilgenbauer, M.; Datta, P.; Qiu, Z.; Mckenzie, S.; Wang, H.; Bowers, D.; Kurokawa, M.; Zheng, Y.-R. Fatty acid-like Pt(iv) prodrugs overcome cisplatin resistance in ovarian cancer by harnessing CD36. Chem. Commun. 2020, 56, 10706–10709. [Google Scholar] [CrossRef]
- Hale, J.S.; Otvos, B.; Sinyuk, M.; Alvarado, A.G.; Hitomi, M.; Stoltz, K.; Wu, Q.; Flavahan, W.; Levison, B.; Johansen, M.L.; et al. Cancer Stem Cell-Specific Scavenger Receptor CD36 Drives Glioblastoma Progression. Stem. Cells 2014, 32, 1746–1758. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhi, Z.; Wang, C.; Xing, H.; Song, G.; Yu, X.; Zhu, Y.; Wang, X.; Zhang, X.; Di, Y. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol. Rep. 2017, 38, 2105–2115. [Google Scholar] [CrossRef] [Green Version]
- Drury, J.; Rychahou, P.G.; He, D.; Jafari, N.; Wang, C.; Lee, E.Y.; Weiss, H.L.; Evers, B.M.; Zaytseva, Y.Y. Inhibition of Fatty Acid Synthase Upregulates Expression of CD36 to Sustain Proliferation of Colorectal Cancer Cells. Front. Oncol. 2020, 10, 1185. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Fan, Z.; Wang, Z.; Dai, Q.; Xiang, Z.; Yuan, F.; Yan, M.; Zhu, Z.; Liu, B.; Li, C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Deng, M.; Cai, X.; Long, L.; Xie, L.; Ma, H.; Zhou, Y.; Liu, S.; Zeng, C. CD36 promotes the epithelial–mesenchymal transition and metastasis in cervical cancer by interacting with TGF-β. J. Transl. Med. 2019, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Su, C.; Luo, X.; Zeng, H.; Zhao, L.; Wei, L.; Zhang, X.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. Dietary oleic acid-induced CD36 promotes cervical cancer cell growth and metastasis via up-regulation Src/ERK pathway. Cancer Lett. 2018, 438, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, O.H.; Córcoles, R.; Fabra, Á.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic Colonization Requires the Repression of the Epithelial-Mesenchymal Transition Inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Pećina-Šlaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.-A.; Kim, S.-H.; Hong, H.K.; Yun, S.H.; Kim, H.C.; Chun, H.-K.; Cho, Y.B.; Lee, W.Y. Loss of E-Cadherin Expression Is Associated with a Poor Prognosis in Stage III Colorectal Cancer. J. Oncol. 2014, 86, 318–328. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christou, N.; Perraud, A.; Blondy, S.; Jauberteau, M.-O.; Battu, S.; Mathonnet, M. E-cadherin: A potential biomarker of colorectal cancer prognosis. Oncol. Lett. 2017, 13, 4571–4576. [Google Scholar] [CrossRef] [Green Version]
- Okugawa, Y.; Toiyama, Y.; Inoue, Y.; Iwata, T.; Fujikawa, H.; Saigusa, S.; Konishi, N.; Tanaka, K.; Uchida, K.; Kusunoki, M. Clinical Significance of Serum Soluble E-cadherin in Colorectal Carcinoma. J. Surg. Res. 2012, 175, e67–e73. [Google Scholar] [CrossRef]
- Wang, R.; Ma, X.; Li, Y.; He, Y.; Huang, D.; Cai, S.; Peng, J. The Characteristics and Prognostic Effect of E-Cadherin Expression in Colorectal Signet Ring Cell Carcinoma. PLoS ONE 2016, 11, e0160527. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zhang, X.; Li, D.; He, P.; Tian, W.; Zeng, B. Expression analysis and clinical significance of eIF4E, VEGF-C, E-cadherin and MMP-2 in colorectal adenocarcinoma. Oncotarget 2016, 7, 85502–85514. [Google Scholar] [CrossRef] [Green Version]
- Kitadai, Y.; Bucana, C.D.; Ellis, L.M.; Anzai, H.; Tahara, E.; Fidler, I.J. In situ mRNA hybridization technique for analysis of metastasis-related genes in human colon carcinoma cells. Am. J. Pathol. 1995, 147, 1238–1247. [Google Scholar]
- Reinhold, W.C.; Reimers, M.A.; Lorenzi, P.; Ho, J.; Shankavaram, U.T.; Ziegler, M.S.; Bussey, K.; Nishizuka, S.; Ikediobi, O.; Pommier, Y.G.; et al. Multifactorial Regulation of E-Cadherin Expression: An Integrative Study. Mol. Cancer Ther. 2010, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31 (Suppl. 1), 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrabec, E.; Naduk, J.; Strek, M.; Hrabec, Z. Type IV collagenases (MMP-2 and MMP-9) and their substrates—Intracellular proteins, hormones, cytokines, chemokines and their receptors. Postępy Biochem. 2007, 53, 37–45. [Google Scholar] [PubMed]
- Illman, S.A.; Lehti, K.; Keski-Oja, J.; Lohi, J. Epilysin (MMP-28) induces TGF-β mediated epithelial to mesenchymal transition in lung carcinoma cells. J. Cell Sci. 2006, 119, 3856–3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, P.; Yanfang, T.; Zhuan, Z.; Jian, W.; Xueming, Z.; Jian, N. MMP28 (epilysin) as a novel promoter of invasion and metastasis in gastric cancer. BMC Cancer 2011, 11, 200. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Pan, Q.; Yan, W.; Wang, Y.; He, X.; Zhao, Z. Overexpression of MMP21 and MMP28 is associated with gastric cancer progression and poor prognosis. Oncol. Lett. 2018, 15, 7776–7782. [Google Scholar] [CrossRef] [PubMed]
- Illman, S.A.; Lohi, J.; Keski-Oja, J. Epilysin (MMP-28)—structure, expression and potential functions. Exp. Dermatol. 2008, 17, 897–907. [Google Scholar] [CrossRef]
- Rychahou, P.; Bae, Y.; Reichel, D.; Zaytseva, Y.Y.; Lee, E.Y.; Napier, D.; Weiss, H.L.; Roller, N.; Frohman, H.; Le, A.-T.; et al. Colorectal cancer lung metastasis treatment with polymer–drug nanoparticles. J. Control. Release 2018, 275, 85–91. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Tseng, W.; Leong, X.; Engleman, E. Orthotopic Mouse Model of Colorectal Cancer. J. Vis. Exp. 2007, 10, e484. [Google Scholar] [CrossRef]
- Agarwal, E.; Brattain, M.G.; Chowdhury, S. Cell survival and metastasis regulation by Akt signaling in colorectal cancer. Cell. Signal. 2013, 25, 1711–1719. [Google Scholar] [CrossRef] [Green Version]
- Elliott, V.A.; Rychahou, P.; Zaytseva, Y.Y.; Evers, B.M. Activation of c-Met and Upregulation of CD44 Expression Are Associated with the Metastatic Phenotype in the Colorectal Cancer Liver Metastasis Model. PLoS ONE 2014, 9, e97432. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 2015, 5, 14752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radisky, E.S.; Radisky, D.C. Matrix Metalloproteinase-Induced Epithelial-Mesenchymal Transition in Breast Cancer. J. Mammary Gland. Biol. Neoplasia 2010, 15, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Lochter, A.; Galosy, S.; Muschler, J.; Freedman, N.; Werb, Z.; Bissell, M.J. Matrix Metalloproteinase Stromelysin-1 Triggers a Cascade of Molecular Alterations That Leads to Stable Epithelial-to-Mesenchymal Conversion and a Premalignant Phenotype in Mammary Epithelial Cells. J. Cell Biol. 1997, 139, 1861–1872. [Google Scholar] [CrossRef]
- McCusker, C.D.; Alfandari, D. Life after proteolysis: Exploring the signaling capabilities of classical cadherin cleavage fragments. Commun. Integr. Biol. 2009, 2, 155–157. [Google Scholar] [CrossRef] [Green Version]
- Ferber, E.C.; Kajita, M.; Wadlow, A.; Tobiansky, L.; Niessen, C.; Ariga, H.; Daniel, J.; Fujita, Y. A Role for the Cleaved Cytoplasmic Domain of E-cadherin in the Nucleus. J. Biol. Chem. 2008, 283, 12691–12700. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Reiss, K.; Ludwig, A.; Buchholz, J.; Scholz, F.; Proksch, E.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and -catenin translocation. Proc. Natl. Acad. Sci. USA 2005, 102, 9182–9187. [Google Scholar] [CrossRef] [Green Version]
- Annett, S.; Moore, G.; Robson, T. Obesity and Cancer Metastasis: Molecular and Translational Perspectives. Cancers 2020, 12, 3798. [Google Scholar] [CrossRef] [PubMed]
- Daquinag, A.C.; Gao, Z.; Fussell, C.; Immaraj, L.; Pasqualini, R.; Arap, W.; Akimzhanov, A.M.; Febbraio, M.; Kolonin, M.G. Fatty acid mobilization from adipose tissue is mediated by CD36 posttranslational modifications and intracellular trafficking. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Xu, S.; Jay, A.; Brunaldi, K.; Huang, N.; Hamilton, J.A. CD36 Enhances Fatty Acid Uptake by Increasing the Rate of Intracellular Esterification but Not Transport across the Plasma Membrane. J. Biochem. 2013, 52, 7254–7261. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.-W.; Wang, J.; Guo, H.; Zhao, Y.-Y.; Sun, H.-H.; Li, Y.-F.; Lai, X.-Y.; Zhao, N.; Wang, X.; Xie, C.; et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Enciu, A.-M.; Radu, E.; Popescu, I.D.; Hinescu, M.E.; Ceafalan, L.C. Targeting CD36 as Biomarker for Metastasis Prognostic: How Far from Translation into Clinical Practice? BioMed Res. Int. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drury, J.; Rychahou, P.G.; Kelson, C.O.; Geisen, M.E.; Wu, Y.; He, D.; Wang, C.; Lee, E.Y.; Evers, B.M.; Zaytseva, Y.Y. Upregulation of CD36, a Fatty Acid Translocase, Promotes Colorectal Cancer Metastasis by Increasing MMP28 and Decreasing E-Cadherin Expression. Cancers 2022, 14, 252. https://doi.org/10.3390/cancers14010252
Drury J, Rychahou PG, Kelson CO, Geisen ME, Wu Y, He D, Wang C, Lee EY, Evers BM, Zaytseva YY. Upregulation of CD36, a Fatty Acid Translocase, Promotes Colorectal Cancer Metastasis by Increasing MMP28 and Decreasing E-Cadherin Expression. Cancers. 2022; 14(1):252. https://doi.org/10.3390/cancers14010252
Chicago/Turabian StyleDrury, James, Piotr G. Rychahou, Courtney O. Kelson, Mariah E. Geisen, Yuanyuan Wu, Daheng He, Chi Wang, Eun Y. Lee, B. Mark Evers, and Yekaterina Y. Zaytseva. 2022. "Upregulation of CD36, a Fatty Acid Translocase, Promotes Colorectal Cancer Metastasis by Increasing MMP28 and Decreasing E-Cadherin Expression" Cancers 14, no. 1: 252. https://doi.org/10.3390/cancers14010252
APA StyleDrury, J., Rychahou, P. G., Kelson, C. O., Geisen, M. E., Wu, Y., He, D., Wang, C., Lee, E. Y., Evers, B. M., & Zaytseva, Y. Y. (2022). Upregulation of CD36, a Fatty Acid Translocase, Promotes Colorectal Cancer Metastasis by Increasing MMP28 and Decreasing E-Cadherin Expression. Cancers, 14(1), 252. https://doi.org/10.3390/cancers14010252