
Targeting the Intrinsic Apoptosis Pathway: A Window of Opportunity for Prostate Cancer

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Cell Death: Multiple Ways to Die
3. The Intrinsic Apoptosis Pathway
4. Evasion of Apoptosis in Cancer
5. Interrogating BCL-2 Protein Dependency
6. Targeting the Intrinsic Apoptosis Pathway with BH3 Mimetics
6.1. Selective BCL-2 Targeting
6.2. Strategies for Selective BCL-XL and Dual BCL-XL/BCL-2 Targeting
6.3. Efforts to Target MCL-1
7. Evasion of Apoptosis in Prostate Cancer and Resistance to Established Therapies
7.1. BCL-2 Proteins in Prostate Cancer
7.2. Resistance to Established Therapies
7.2.1. Resistance to ADT
7.2.2. Resistance to AR Signaling Inhibition
7.2.3. Resistance to Chemotherapy
8. Targeting the Intrinsic Apoptosis Pathway in Prostate Cancer with BH3 Mimetics
9. Future Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Glocal Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 23 March 2021).
- Siegel, D.A.; O’Neil, M.E.; Richards, T.B.; Dowling, N.F.; Weir, H.K. Prostate cancer incidence and survival, by stage and race/ethnicity—United States, 2001–2017. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1473–1480. [Google Scholar] [CrossRef]
- Stephenson, A.J.; Scardino, P.T.; Eastham, J.A.; Bianco, F.J., Jr.; Dotan, Z.A.; Fearn, P.A.; Kattan, M.W. Preoperative nomogram predicting the 10-year probability of prostate cancer recurrence after radical prostatectomy. J. Natl. Cancer Inst. 2006, 98, 715–717. [Google Scholar] [CrossRef] [Green Version]
- Hull, G.W.; Rabbani, F.; Abbas, F.; Wheeler, T.M.; Kattan, M.W.; Scardino, P.T. Cancer control with radical prostatectomy alone in 1000 consecutive patients. J. Urol. 2002, 167, 528–534. [Google Scholar] [CrossRef]
- Kupelian, P.; Katcher, J.; Levin, H.; Zippe, C.; Klein, E. Correlation of clinical and pathologic factors with rising prostate-specific antigen profiles after radical prostatectomy alone for clinically localized prostate cancer. Urology 1996, 48, 249–260. [Google Scholar] [CrossRef]
- Kupelian, P.A.; Mahadevan, A.; Reddy, C.A.; Reuther, A.M.; Klein, E.A. Use of different definitions of biochemical failure after external beam radiotherapy changes conclusions about relative treatment efficacy for localized prostate cancer. Urology 2006, 68, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Wilding, G. The importance of steroid hormones in prostate cancer. Cancer Surv. 1992, 14, 113–130. [Google Scholar] [PubMed]
- Sartor, O.; de Bono, J.S. Metastatic prostate cancer. N. Engl. J. Med. 2018, 378, 1653–1654. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Huggins, C.; Stevens, R.E.; Hodges, C.V. Studies on prostatic cancer: II. The effects of castration on advanced carcinoma of the prostate gland. Arch. Surg. 1941, 43, 645–657. [Google Scholar] [CrossRef]
- Westaby, D.; Maza, M.; Paschalis, A.; Jimenez-Vacas, J.M.; Welti, J.; de Bono, J.; Sharp, A. A new old target: Androgen receptor signaling and advanced prostate cancer. Annu. Rev. Pharmacol. Toxicol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Mehtala, J.; Zong, J.; Vassilev, Z.; Brobert, G.; Gabarro, M.S.; Stattin, P.; Khanfir, H. Overall survival and second primary malignancies in men with metastatic prostate cancer. PLoS ONE 2020, 15, e0227552. [Google Scholar] [CrossRef] [Green Version]
- Moreira, D.M.; Howard, L.E.; Sourbeer, K.N.; Amarasekara, H.S.; Chow, L.C.; Cockrell, D.C.; Pratson, C.L.; Hanyok, B.T.; Aronson, W.J.; Kane, C.J.; et al. Predicting time from metastasis to overall survival in castration-resistant prostate cancer: Results from SEARCH. Clin. Genitourin. Cancer 2017, 15, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N., Jr.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [Green Version]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Fizazi, K.; Scher, H.I.; Molina, A.; Logothetis, C.J.; Chi, K.N.; Jones, R.J.; Staffurth, J.N.; North, S.; Vogelzang, N.J.; Saad, F.; et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012, 13, 983–992. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.; Sternberg, C.N.; Miller, K.; Logothetis, C.J.; Shore, N.D.; Small, E.J.; et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015, 16, 152–160. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Evans, C.P.; Kim, C.S.; Kimura, G.; et al. Enzalutamide in men with chemotherapy-naive metastatic castration-resistant prostate cancer: Extended analysis of the phase 3 PREVAIL study. Eur. Urol. 2017, 71, 151–154. [Google Scholar] [CrossRef] [Green Version]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A randomized, phase III study of androgen deprivation therapy with enzalutamide or placebo in men with metastatic hormone-sensitive prostate cancer. J. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Juarez Soto, A.; Merseburger, A.S.; Ozguroglu, M.; Uemura, H.; et al. Apalutamide for metastatic, castration-sensitive prostate cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with standard first-line therapy in metastatic prostate cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef]
- Tucci, M.; Bertaglia, V.; Vignani, F.; Buttigliero, C.; Fiori, C.; Porpiglia, F.; Scagliotti, G.V.; Di Maio, M. Addition of docetaxel to androgen deprivation therapy for patients with hormone-sensitive metastatic prostate cancer: A systematic review and meta-analysis. Eur. Urol. 2016, 69, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): Final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- James, N.D.; De Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. New Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Bumbaca, B.; Li, W. Taxane resistance in castration-resistant prostate cancer: Mechanisms and therapeutic strategies. Acta Pharm. Sin. B 2018, 8, 518–529. [Google Scholar] [CrossRef]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- De Bono, J.S.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Gafanov, R.; et al. LBA4 IPATential150: Phase III study of ipatasertib (ipat) plus abiraterone (abi) vs placebo (pbo) plus abi in metastatic castration-resistant prostate cancer (mCRPC). Ann. Oncol. 2020, 31, S1153–S1154. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweat, S.D.; Pacelli, A.; Murphy, G.P.; Bostwick, D.G. Prostate-specific membrane antigen expression is greatest in prostate adenocarcinoma and lymph node metastases. Urology 1998, 52, 637–640. [Google Scholar] [CrossRef]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Kramer, G.; Schwarz, S.; Hagg, M.; Havelka, A.M.; Linder, S. Docetaxel induces apoptosis in hormone refractory prostate carcinomas during multiple treatment cycles. Br. J. Cancer 2006, 94, 1592–1598. [Google Scholar] [CrossRef]
- Zhang, K.X.; Firus, J.; Prieur, B.; Jia, W.; Rennie, P.S. To die or to survive, a fatal question for the destiny of prostate cancer cells after androgen deprivation therapy. Cancers 2011, 3, 1498–1512. [Google Scholar] [CrossRef] [Green Version]
- Westin, P.; Stattin, P.; Damber, J.E.; Bergh, A. Castration therapy rapidly induces apoptosis in a minority and decreases cell proliferation in a majority of human prostatic tumors. Am. J. Pathol. 1995, 146, 1368–1375. [Google Scholar] [PubMed]
- Ohlson, N.; Wikstrom, P.; Stattin, P.; Bergh, A. Cell proliferation and apoptosis in prostate tumors and adjacent non-malignant prostate tissue in patients at different time-points after castration treatment. Prostate 2005, 62, 307–315. [Google Scholar] [CrossRef]
- Agus, D.B.; Cordon-Cardo, C.; Fox, W.; Drobnjak, M.; Koff, A.; Golde, D.W.; Scher, H.I. Prostate cancer cell cycle regulators: Response to androgen withdrawal and development of androgen independence. J. Natl. Cancer Inst. 1999, 91, 1869–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, D.G.; Giribaldi, M.G.; Munoz, A.; Halvorsen, K.; Patel, A.; Jorda, M.; Perez-Stable, C.; Rai, P. Androgen deprivation-induced senescence promotes outgrowth of androgen-refractory prostate cancer cells. PLoS ONE 2013, 8, e68003. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Castilla, C.; Congregado, B.; Chinchon, D.; Torrubia, F.J.; Japon, M.A.; Saez, C. Bcl-xL is overexpressed in hormone-resistant prostate cancer and promotes survival of LNCaP cells via interaction with proapoptotic Bak. Endocrinology 2006, 147, 4960–4967. [Google Scholar] [CrossRef]
- Handle, F.; Prekovic, S.; Helsen, C.; Van den Broeck, T.; Smeets, E.; Moris, L.; Eerlings, R.; Kharraz, S.E.; Urbanucci, A.; Mills, I.G.; et al. Drivers of AR indifferent anti-androgen resistance in prostate cancer cells. Sci. Rep. 2019, 9, 13786. [Google Scholar] [CrossRef] [Green Version]
- Raffo, A.J.; Perlman, H.; Chen, M.W.; Day, M.L.; Streitman, J.S.; Buttyan, R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995, 55, 4438–4445. [Google Scholar]
- Kim, J.H.; Lee, H.; Shin, E.A.; Kim, D.H.; Choi, J.B.; Kim, S.H. Implications of Bcl-2 and its interplay with other molecules and signaling pathways in prostate cancer progression. Expert Opin. Ther. Targets 2017, 21, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Fukuchi, J.; Hiipakka, R.A.; Kokontis, J.M.; Xiang, J. Up-regulation of Bcl-2 is required for the progression of prostate cancer cells from an androgen-dependent to an androgen-independent growth stage. Cell Res. 2007, 17, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, I.; Rando, R.; Ojwang, J.; Cossum, P.; Stein, C.A. Bcl-xL in prostate cancer cells: Effects of overexpression and down-regulation on chemosensitivity. Cancer Res. 2000, 60, 6052–6060. [Google Scholar]
- Reiner, T.; de Las Pozas, A.; Parrondo, R.; Palenzuela, D.; Cayuso, W.; Rai, P.; Perez-Stable, C. Mcl-1 protects prostate cancer cells from cell death mediated by chemotherapy-induced DNA damage. Oncoscience 2015, 2, 703–715. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.J.; Leung, H.Y. Evasion of cell death: A contributory factor in prostate cancer development and treatment resistance. Cancer Lett. 2021, 520, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Maiuri, M.C.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell death modalities: Classification and pathophysiological implications. Cell Death Differ. 2007, 14, 1237–1243. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kepp, O.; Kroemer, G. Regulated cell death and adaptive stress responses. Cell Mol. Life Sci. 2016, 73, 2405–2410. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatim, N.; Cullen, S.; Albert, M.L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef]
- Strasser, A.; Harris, A.W.; Huang, D.C.; Krammer, P.H.; Cory, S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 1995, 14, 6136–6147. [Google Scholar] [CrossRef]
- Barnhart, B.C.; Alappat, E.C.; Peter, M.E. The CD95 type I/type II model. Semin. Immunol. 2003, 15, 185–193. [Google Scholar] [CrossRef]
- Ozoren, N.; El-Deiry, W.S. Defining characteristics of types I and II apoptotic cells in response to TRAIL. Neoplasia 2002, 4, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.M.; Wang, K.; Gross, A.; Zhao, Y.; Zinkel, S.; Klocke, B.; Roth, K.A.; Korsmeyer, S.J. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999, 400, 886–891. [Google Scholar] [CrossRef]
- Wang, K.J.; Wang, K.Y.; Zhang, H.Z.; Meng, X.Y.; Chen, J.F.; Wang, P.; Jiang, J.H.; Ma, Q. Up-regulation of RIP3 alleviates prostate cancer progression by activation of RIP3/MLKL signaling pathway and induction of necroptosis. Front. Oncol. 2020, 10, 1720. [Google Scholar] [CrossRef]
- Ghoochani, A.; Hsu, E.C.; Aslan, M.; Rice, M.A.; Nguyen, H.M.; Brooks, J.D.; Corey, E.; Paulmurugan, R.; Stoyanova, T. Ferroptosis inducers are a novel therapeutic approach for advanced prostate cancer. Cancer Res. 2021, 81, 1583–1594. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Ni Chonghaile, T.; Sarosiek, K.A.; Vo, T.T.; Ryan, J.A.; Tammareddi, A.; Moore Vdel, G.; Deng, J.; Anderson, K.C.; Richardson, P.; Tai, Y.T.; et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 2011, 334, 1129–1133. [Google Scholar] [CrossRef] [Green Version]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Huang, K.; O’Neill, K.L.; Li, J.; Zhou, W.; Han, N.; Pang, X.; Wu, W.; Struble, L.; Borgstahl, G.; Liu, Z.; et al. BH3-only proteins target BCL-xL/MCL-1, not BAX/BAK, to initiate apoptosis. Cell Res. 2019, 29, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Uren, R.T.; Dengler, M.A.; Shi, M.X.; Uno, E.; Adams, J.M.; Dewson, G.; Kluck, R.M. Robust autoactivation for apoptosis by BAK but not BAX highlights BAK as an important therapeutic target. Cell Death Dis. 2020, 11, 268. [Google Scholar] [CrossRef] [PubMed]
- Greaves, G.; Milani, M.; Butterworth, M.; Carter, R.J.; Byrne, D.P.; Eyers, P.A.; Luo, X.; Cohen, G.M.; Varadarajan, S. BH3-only proteins are dispensable for apoptosis induced by pharmacological inhibition of both MCL-1 and BCL-XL. Cell Death Differ. 2019, 26, 1037–1047. [Google Scholar] [CrossRef]
- O’Neill, K.L.; Huang, K.; Zhang, J.; Chen, Y.; Luo, X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016, 30, 973–988. [Google Scholar] [CrossRef] [Green Version]
- Silke, J.; Meier, P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008730. [Google Scholar] [CrossRef]
- Marsden, V.S.; O’Connor, L.; O’Reilly, L.A.; Silke, J.; Metcalf, D.; Ekert, P.G.; Huang, D.C.; Cecconi, F.; Kuida, K.; Tomaselli, K.J.; et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 2002, 419, 634–637. [Google Scholar] [CrossRef]
- Ichim, G.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, J.S.; Mason, S.M.; Athineos, D.; et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Sarosiek, K.A.; Fraser, C.; Muthalagu, N.; Bhola, P.D.; Chang, W.; McBrayer, S.K.; Cantlon, A.; Fisch, S.; Golomb-Mello, G.; Ryan, J.A.; et al. Developmental regulation of mitochondrial apoptosis by c-Myc governs age- and tissue-specific sensitivity to cancer therapeutics. Cancer Cell 2017, 31, 142–156. [Google Scholar] [CrossRef] [Green Version]
- Madden, S.D.; Donovan, M.; Cotter, T.G. Key apoptosis regulating proteins are down-regulated during postnatal tissue development. Int. J. Dev. Biol. 2007, 51, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.; Tait, S.W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to inhibit Myc and their clinical applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, S.; Cao, S.; Jin, L.; Kobelski, M.; Schouest, B.; Wang, X.; Ungerleider, N.; Baddoo, M.; Zhang, W.; Corey, E.; et al. A positive role of c-Myc in regulating androgen receptor and its splice variants in prostate cancer. Oncogene 2019, 38, 4977–4989. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Woods, N.T.; Yamaguchi, H.; Lee, F.Y.; Bhalla, K.N.; Wang, H.G. Anoikis, initiated by Mcl-1 degradation and Bim induction, is deregulated during oncogenesis. Cancer Res. 2007, 67, 10744–10752. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Eguchi, Y.; Kosaka, H.; Kamiike, W.; Matsuda, H.; Tsujimoto, Y. Prevention of hypoxia-induced cell death by Bcl-2 and Bcl-xL. Nature 1995, 374, 811–813. [Google Scholar] [CrossRef]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef] [PubMed]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef] [Green Version]
- Inuzuka, H.; Shaik, S.; Onoyama, I.; Gao, D.; Tseng, A.; Maser, R.S.; Zhai, B.; Wan, L.; Gutierrez, A.; Lau, A.W.; et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 2011, 471, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; He, X.; Hsu, J.M.; Xia, W.; Chen, C.T.; Li, L.Y.; Lee, D.F.; Liu, J.C.; Zhong, Q.; Wang, X.; et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell Biol. 2007, 27, 4006–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwickart, M.; Huang, X.; Lill, J.R.; Liu, J.; Ferrando, R.; French, D.M.; Maecker, H.; O’Rourke, K.; Bazan, F.; Eastham-Anderson, J.; et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010, 463, 103–107. [Google Scholar] [CrossRef]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef] [Green Version]
- Morales, A.A.; Kurtoglu, M.; Matulis, S.M.; Liu, J.; Siefker, D.; Gutman, D.M.; Kaufman, J.L.; Lee, K.P.; Lonial, S.; Boise, L.H. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood 2011, 118, 1329–1339. [Google Scholar] [CrossRef]
- Weeden, C.E.; Ah-Cann, C.; Holik, A.Z.; Pasquet, J.; Garnier, J.M.; Merino, D.; Lessene, G.; Asselin-Labat, M.L. Dual inhibition of BCL-XL and MCL-1 is required to induce tumour regression in lung squamous cell carcinomas sensitive to FGFR inhibition. Oncogene 2018, 37, 4475–4488. [Google Scholar] [CrossRef]
- Soderquist, R.S.; Crawford, L.; Liu, E.; Lu, M.; Agarwal, A.; Anderson, G.R.; Lin, K.H.; Winter, P.S.; Cakir, M.; Wood, K.C. Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nat. Commun. 2018, 9, 3513. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.; Letai, A. BH3 profiling in whole cells by fluorimeter or FACS. Methods 2013, 61, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.N.; Khong, T.; Segal, D.; Yao, Y.; Riffkin, C.D.; Garnier, J.M.; Khaw, S.L.; Lessene, G.; Spencer, A.; Herold, M.J.; et al. Hierarchy for targeting prosurvival BCL2 family proteins in multiple myeloma: Pivotal role of MCL1. Blood 2016, 128, 1834–1844. [Google Scholar] [CrossRef] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a cancer dependency map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Kelly, W.K.; Wilding, G.; Leopold, L.; Brill, K.; Somer, B. An open-label, multicenter, phase I/II study of single-agent AT-101 in men with castrate-resistant prostate cancer. Clin. Cancer Res. 2009, 15, 3172–3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmer, A.D.; Raza, A.; Carter, T.H.; Claxton, D.; Erba, H.; DeAngelo, D.J.; Tallman, M.S.; Goard, C.; Borthakur, G. A multicenter phase I/II study of obatoclax mesylate administered as a 3- or 24-hour infusion in older patients with previously untreated acute myeloid leukemia. PLoS ONE 2014, 9, e108694. [Google Scholar] [CrossRef] [PubMed]
- Besbes, S.; Mirshahi, M.; Pocard, M.; Billard, C. New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget 2015, 6, 12862–12871. [Google Scholar] [CrossRef] [Green Version]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, L.; Camidge, D.R.; Ribeiro de Oliveira, M.; Bonomi, P.; Gandara, D.; Khaira, D.; Hann, C.L.; McKeegan, E.M.; Litvinovich, E.; Hemken, P.M.; et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J. Clin. Oncol. 2011, 29, 909–916. [Google Scholar] [CrossRef] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Montesinos, P.; Ivanov, V.; DiNardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Gerecitano, J.; Robak, T.; De la Serna, J.; et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N. Engl. J. Med. 2018, 378, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Al-Sawaf, O.; Bahlo, J.; Fink, A.M.; Tandon, M.; Dixon, M.; Robrecht, S.; Warburton, S.; Humphrey, K.; Samoylova, O.; et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N. Engl. J. Med. 2019, 380, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Ma, S.; Kipps, T.J.; Coutre, S.E.; Davids, M.S.; Eichhorst, B.; Hallek, M.; Byrd, J.C.; Humphrey, K.; Zhou, L.; et al. Efficacy of venetoclax in relapsed chronic lymphocytic leukemia is influenced by disease and response variables. Blood 2019, 134, 111–122. [Google Scholar] [CrossRef]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Klanova, M.; Andera, L.; Brazina, J.; Svadlenka, J.; Benesova, S.; Soukup, J.; Prukova, D.; Vejmelkova, D.; Jaksa, R.; Helman, K.; et al. Targeting of BCL2 family proteins with ABT-199 and homoharringtonine reveals BCL2- and MCL1-dependent subgroups of diffuse large B-Cell lymphoma. Clin. Cancer Res. 2016, 22, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, M.; Nkwocha, J.; Hawkins, E.; Pei, X.; Parker, R.E.; Kmieciak, M.; Leverson, J.D.; Sampath, D.; Ferreira-Gonzalez, A.; Grant, S. Cotargeting BCL-2 and PI3K induces BAX-dependent mitochondrial apoptosis in AML cells. Cancer Res. 2018, 78, 3075–3086. [Google Scholar] [CrossRef] [Green Version]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in cancer: Advances, challenges, and perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef] [PubMed]
- Lasica, M.; Anderson, M.A. Review of venetoclax in CLL, AML and multiple myeloma. J. Pers. Med. 2021, 11, 463. [Google Scholar] [CrossRef]
- Guieze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernandez-Sanchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial reprogramming underlies resistance to BCL-2 inhibition in lymphoid malignancies. Cancer Cell 2019, 36, 369–384. [Google Scholar] [CrossRef]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; d’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Lochmann, T.L.; Floros, K.V.; Naseri, M.; Powell, K.M.; Cook, W.; March, R.J.; Stein, G.T.; Greninger, P.; Maves, Y.K.; Saunders, L.R.; et al. Venetoclax is effective in small-cell lung cancers with high BCL-2 expression. Clin. Cancer Res. 2018, 24, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Krump, N.A.; Herlyn, M.; You, J. Combining DNA damage induction with BCL-2 inhibition to enhance Merkel cell carcinoma cytotoxicity. Biology 2020, 9, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittle, J.R.; Vaillant, F.; Surgenor, E.; Policheni, A.N.; Giner, G.; Capaldo, B.D.; Chen, H.R.; Liu, H.K.; Dekkers, J.F.; Sachs, N.; et al. Dual targeting of CDK4/6 and BCL2 pathways augments tumor response in estrogen receptor-positive breast cancer. Clin. Cancer Res. 2020, 26, 4120–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpel-Massler, G.; Ishida, C.T.; Bianchetti, E.; Shu, C.; Perez-Lorenzo, R.; Horst, B.; Banu, M.; Roth, K.A.; Bruce, J.N.; Canoll, P.; et al. Inhibition of mitochondrial matrix chaperones and antiapoptotic Bcl-2 family proteins empower antitumor therapeutic responses. Cancer Res. 2017, 77, 3513–3526. [Google Scholar] [CrossRef] [Green Version]
- Lok, S.W.; Whittle, J.R.; Vaillant, F.; Teh, C.E.; Lo, L.L.; Policheni, A.N.; Bergin, A.R.T.; Desai, J.; Ftouni, S.; Gandolfo, L.C.; et al. A phase Ib dose-escalation and expansion study of the BCL2 inhibitor venetoclax combined with tamoxifen in ER and BCL2-positive metastatic breast cancer. Cancer Discov. 2019, 9, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, G.J.; Bowen, R.; Jerzak, K.J.; Song, X.; Decker, T.; Boyle, F.M.; McCune, S.L.; Armstrong, A.; Shannon, C.M.; Bertelli, G.; et al. Results from VERONICA: A randomized, phase II study of second-/third-line venetoclax (VEN) + fulvestrant (F) versus F alone in estrogen receptor (ER)-positive, HER2-negative, locally advanced, or metastatic breast cancer (LA/MBC). J. Clin. Oncol. 2021, 39, 1004. [Google Scholar] [CrossRef]
- Kehr, S.; Vogler, M. It’s time to die: BH3 mimetics in solid tumors. Biochimica Biophysica Acta Mol. Cell. Res. 2021, 1868, 118987. [Google Scholar] [CrossRef]
- Ow, T.J.; Fulcher, C.D.; Thomas, C.; Broin, P.O.; Lopez, A.; Reyna, D.E.; Smith, R.V.; Sarta, C.; Prystowsky, M.B.; Schlecht, N.F.; et al. Optimal targeting of BCL-family proteins in head and neck squamous cell carcinoma requires inhibition of both BCL-xL and MCL-1. Oncotarget 2019, 10, 494–510. [Google Scholar] [CrossRef]
- Levesley, J.; Steele, L.; Bruning-Richardson, A.; Davison, A.; Zhou, J.; Ding, C.; Lawler, S.; Short, S.C. Selective BCL-XL inhibition promotes apoptosis in combination with MLN8237 in medulloblastoma and pediatric glioblastoma cells. Neuro. Oncol. 2018, 20, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.F.; Harris, T.J.; Tran, S.; Evangelista, M.; Arulananda, S.; John, T.; Ramnac, C.; Hobbs, C.; Zhu, H.; Gunasingh, G.; et al. BCL-XL and MCL-1 are the key BCL-2 family proteins in melanoma cell survival. Cell Death Dis. 2019, 10, 342. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Amundson, S.A.; Myers, T.G.; Scudiero, D.; Kitada, S.; Reed, J.C.; Fornace, A.J., Jr. An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Res. 2000, 60, 6101–6110. [Google Scholar] [PubMed]
- Trisciuoglio, D.; Tupone, M.G.; Desideri, M.; Di Martile, M.; Gabellini, C.; Buglioni, S.; Pallocca, M.; Alessandrini, G.; D’Aguanno, S.; Del Bufalo, D. BCL-XL overexpression promotes tumor progression-associated properties. Cell Death Dis. 2017, 8, 3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranski, Z.; de Jong, Y.; Ilkova, T.; Peterse, E.F.; Cleton-Jansen, A.M.; van de Water, B.; Hogendoorn, P.C.; Bovee, J.V.; Danen, E.H. Pharmacological inhibition of Bcl-xL sensitizes osteosarcoma to doxorubicin. Oncotarget 2015, 6, 36113–36125. [Google Scholar] [CrossRef] [Green Version]
- Abed, M.N.; Abdullah, M.I.; Richardson, A. Antagonism of Bcl-XL is necessary for synergy between carboplatin and BH3 mimetics in ovarian cancer cells. J. Ovarian Res. 2016, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Lucantoni, F.; Lindner, A.U.; O’Donovan, N.; Dussmann, H.; Prehn, J.H.M. Systems modeling accurately predicts responses to genotoxic agents and their synergism with BCL-2 inhibitors in triple negative breast cancer cells. Cell Death Dis. 2018, 9, 42. [Google Scholar] [CrossRef]
- Tao, Z.F.; Hasvold, L.; Wang, L.; Wang, X.; Petros, A.M.; Park, C.H.; Boghaert, E.R.; Catron, N.D.; Chen, J.; Colman, P.M.; et al. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med. Chem. Lett. 2014, 5, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Doherty, G.A.; Judd, A.S.; Tao, Z.F.; Hansen, T.M.; Frey, R.R.; Song, X.; Bruncko, M.; Kunzer, A.R.; Wang, X.; et al. Discovery of A-1331852, a first-in-class, potent, and orally-bioavailable BCL-XL inhibitor. ACS Med. Chem. Lett. 2020, 11, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, 279ra40. [Google Scholar] [CrossRef]
- Lessene, G.; Czabotar, P.E.; Sleebs, B.E.; Zobel, K.; Lowes, K.N.; Adams, J.M.; Baell, J.B.; Colman, P.M.; Deshayes, K.; Fairbrother, W.J.; et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013, 9, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Carneiro, B.A.; Dowlati, A.; Razak, A.R.A.; Chae, Y.K.; Villella, J.A.; Coppola, S.; Englert, S.; Phillips, A.C.; Souers, A.J.; et al. A first-in-human study of mirzotamab clezutoclax as monotherapy and in combination with taxane therapy in relapsed/refractory solid tumors: Dose escalation results. J. Clin. Oncol. 2021, 39, 3015. [Google Scholar] [CrossRef]
- He, Y.; Koch, R.; Budamagunta, V.; Zhang, P.; Zhang, X.; Khan, S.; Thummuri, D.; Ortiz, Y.T.; Zhang, X.; Lv, D.; et al. DT2216-a Bcl-xL-specific degrader is highly active against Bcl-xL-dependent T cell lymphomas. J. Hematol. Oncol. 2020, 13, 95. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Kolb, R.; De, U.; Khan, S.; Luo, Y.; Kim, M.C.; Yu, H.; Wu, C.; Mo, J.; Zhang, X.; Zhang, P.; et al. Proteolysis-targeting chimera against BCL-XL destroys tumor-infiltrating regulatory T cells. Nat. Commun. 2021, 12, 1281. [Google Scholar] [CrossRef]
- Patterson, C.M.; Balachander, S.B.; Grant, I.; Pop-Damkov, P.; Kelly, B.; McCoull, W.; Parker, J.; Giannis, M.; Hill, K.J.; Gibbons, F.D.; et al. Design and optimisation of dendrimer-conjugated Bcl-2/xL inhibitor, AZD0466, with improved therapeutic index for cancer therapy. Commun. Biol. 2021, 4, 112. [Google Scholar] [CrossRef]
- Arulananda, S.; O’Brien, M.; Evangelista, M.; Jenkins, L.J.; Poh, A.R.; Walkiewicz, M.; Leong, T.; Mariadason, J.M.; Cebon, J.; Balachander, S.B.; et al. A novel BH3-mimetic, AZD0466, targeting BCL-XL and BCL-2 is effective in pre-clinical models of malignant pleural mesothelioma. Cell Death Discov. 2021, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, N.J.; Rasco, D.W.; Zeng, Q.; Tang, Y.; Liang, Z.; Wang, H.; Lu, M.; Chen, J.; Fu, L.; Wang, C.; et al. First-in-human study of palcitoclax (APG-1252), a novel dual Bcl-2/Bcl-xL inhibitor, demonstrated advantages in platelet safety while maintaining anticancer effect in U.S. patients with metastatic solid tumors. J. Clin. Oncol. 2020, 38, 3509. [Google Scholar] [CrossRef]
- Hamilton, C.; Fox, J.P.; Longley, D.B.; Higgins, C.A. Therapeutics targeting the core apoptotic machinery. Cancers 2021, 13, 2618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gojo, I.; Fenton, R.G. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood 2002, 99, 1885–1893. [Google Scholar] [CrossRef]
- Perciavalle, R.M.; Opferman, J.T. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol. 2013, 23, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabow, S.; Delbridge, A.R.; Valente, L.J.; Strasser, A. MCL-1 but not BCL-XL is critical for the development and sustained expansion of thymic lymphoma in p53-deficient mice. Blood 2014, 124, 3939–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; Herold, M.J.; et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Bolomsky, A.; Vogler, M.; Kose, M.C.; Heckman, C.A.; Ehx, G.; Ludwig, H.; Caers, J. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J. Hematol. Oncol. 2020, 13, 173. [Google Scholar] [CrossRef]
- Williams, M.M.; Elion, D.L.; Rahman, B.; Hicks, D.J.; Sanchez, V.; Cook, R.S. Therapeutic inhibition of Mcl-1 blocks cell survival in estrogen receptor-positive breast cancers. Oncotarget 2019, 10, 5389–5402. [Google Scholar] [CrossRef] [Green Version]
- Bhagwat, N.; Grego, A.; Gowen-MacDonald, W.; Wang, M.; Cowart, M.; Wu, X.; Zhuo, J.; Combs, A.; Ruggeri, B.; Scherle, P.; et al. Abstract 983: Preclinical characterization of PRT1419, a potent, selective and orally available inhibitor of MCL1. Cancer Res. 2021, 81, 983. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef]
- Caenepeel, S.; Karen, R.; Belmontes, B.; Verlinsky, A.; Tan, H.; Yang, Y.; Chen, X.; Li, K.; Allen, J.; Wahlstrom, J.; et al. Abstract 6218: Discovery and preclinical evaluation of AMG 397, a potent, selective and orally bioavailable MCL1 inhibitor. Cancer Res. 2020, 80, 6218. [Google Scholar] [CrossRef]
- Caenepeel, S.; Brown, S.P.; Belmontes, B.; Moody, G.; Keegan, K.S.; Chui, D.; Whittington, D.A.; Huang, X.; Poppe, L.; Cheng, A.C.; et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018, 8, 1582–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slomp, A.; Moesbergen, L.M.; Gong, J.N.; Cuenca, M.; von dem Borne, P.A.; Sonneveld, P.; Huang, D.C.S.; Minnema, M.C.; Peperzak, V. Multiple myeloma with 1q21 amplification is highly sensitive to MCL-1 targeting. Blood Adv. 2019, 3, 4202–4214. [Google Scholar] [CrossRef]
- Yasuda, Y.; Ozasa, H.; Kim, Y.H.; Yamazoe, M.; Ajimizu, H.; Yamamoto Funazo, T.; Nomizo, T.; Tsuji, T.; Yoshida, H.; Sakamori, Y.; et al. MCL1 inhibition is effective against a subset of small-cell lung cancer with high MCL1 and low BCL-XL expression. Cell Death Dis. 2020, 11, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munkhbaatar, E.; Dietzen, M.; Agrawal, D.; Anton, M.; Jesinghaus, M.; Boxberg, M.; Pfarr, N.; Bidola, P.; Uhrig, S.; Hockendorf, U.; et al. MCL-1 gains occur with high frequency in lung adenocarcinoma and can be targeted therapeutically. Nat. Commun. 2020, 11, 4527. [Google Scholar] [CrossRef]
- Sale, M.J.; Minihane, E.; Monks, N.R.; Gilley, R.; Richards, F.M.; Schifferli, K.P.; Andersen, C.L.; Davies, E.J.; Vicente, M.A.; Ozono, E.; et al. Targeting melanoma’s MCL1 bias unleashes the apoptotic potential of BRAF and ERK1/2 pathway inhibitors. Nat. Commun. 2019, 10, 5167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallet, S.; Fan, F.; Malvestiti, S.; Pecherstorfer, M.; Sattler, M.; Schneeweiss, A.; Schulze-Bergkamen, H.; Opferman, J.T.; Cardone, M.H.; Jager, D.; et al. Rationally derived drug combinations with the novel Mcl-1 inhibitor EU-5346 in breast cancer. Breast Cancer Res. Treat. 2019, 173, 585–596. [Google Scholar] [CrossRef]
- Merino, D.; Whittle, J.R.; Vaillant, F.; Serrano, A.; Gong, J.N.; Giner, G.; Maragno, A.L.; Chanrion, M.; Schneider, E.; Pal, B.; et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, A.; Rosenberg, A.S.; Jakubowiak, A.; Raje, N.; Chatterjee, M.; Trudel, S.; Bahlis, N.J.; Siegel, D.S.; Wilop, S.; Harrison, S.J.; et al. A phase 1, first-in-human study of AMG 176, a selective MCL-1 inhibitor, in patients with relapsed or refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, e53–e54. [Google Scholar] [CrossRef]
- Wang, X.; Bathina, M.; Lynch, J.; Koss, B.; Calabrese, C.; Frase, S.; Schuetz, J.D.; Rehg, J.E.; Opferman, J.T. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013, 27, 1351–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.L.; Roberts, D.J.; Kubli, D.A.; Lee, Y.; Quinsay, M.N.; Owens, J.B.; Fischer, K.M.; Sussman, M.A.; Miyamoto, S.; Gustafsson, A.B. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013, 27, 1365–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushima, H.; Hosaka, Y.; Suzuki, M.; Mizutani, T.; Ishizuka, H.; Kawabe, K. bl-2 [corrected] expression on prostate cancer and its relationship to cell cycle and prognosis. Int. J. Urol. 1996, 3, 113–117. [Google Scholar] [CrossRef]
- Krajewska, M.; Krajewski, S.; Epstein, J.I.; Shabaik, A.; Sauvageot, J.; Song, K.; Kitada, S.; Reed, J.C. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am. J. Pathol. 1996, 148, 1567–1576. [Google Scholar]
- Yoshino, T.; Shiina, H.; Urakami, S.; Kikuno, N.; Yoneda, T.; Shigeno, K.; Igawa, M. Bcl-2 expression as a predictive marker of hormone-refractory prostate cancer treated with taxane-based chemotherapy. Clin. Cancer Res. 2006, 12, 6116–6124. [Google Scholar] [CrossRef] [Green Version]
- Colombel, M.; Symmans, F.; Gil, S.; O’Toole, K.M.; Chopin, D.; Benson, M.; Olsson, C.A.; Korsmeyer, S.; Buttyan, R. Detection of the apoptosis-suppressing oncoprotein bc1-2 in hormone-refractory human prostate cancers. Am. J. Pathol. 1993, 143, 390–400. [Google Scholar] [PubMed]
- Anvari, K.; Seilanian Toussi, M.; Kalantari, M.; Naseri, S.; Karimi Shahri, M.; Ahmadnia, H.; Katebi, M.; Sedighi Pashaki, A.; Dayani, M.; Broumand, M. Expression of Bcl-2 and Bax in advanced or metastatic prostate carcinoma. Urol. J. 2012, 9, 381–388. [Google Scholar]
- Cho, I.C.; Chung, H.S.; Cho, K.S.; Kim, J.E.; Joung, J.Y.; Seo, H.K.; Chung, J.; Park, W.S.; Hong, E.K.; Lee, K.H. Bcl-2 as a predictive factor for biochemical recurrence after radical prostatectomy: An interim analysis. Cancer Res. Treat. 2010, 42, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Kallakury, B.S.; Sheehan, C.E.; Fisher, H.A.; Kaufman, R.P., Jr.; Ross, J.S. Survivin and Bcl-2 expression in prostatic adenocarcinomas. Arch. Pathol. Lab. Med. 2004, 128, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Borre, M.; Stausbol-Gron, B.; Nerstrom, B.; Overgaard, J. Immunohistochemical BCL-2 and KI-67 expression predict survival in prostate cancer patients followed expectantly. Prostate Cancer Prostatic Dis. 1998, 1, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Pfeil, K.; Eder, I.E.; Bektic, J.; Huebl, H.; Pycha, A.; Schaefer, G.; Rogatsch, H.; Bartsch, G.; Klocker, H. The anti-apoptotic protein Mcl-1 is overexpressed in prostate cancer. Cancer Res. 2004, 64, 1193. [Google Scholar]
- Liang, Y.; Jeganathan, S.; Marastoni, S.; Sharp, A.; Figueiredo, I.; Marcellus, R.; Mawson, A.; Shalev, Z.; Pesic, A.; Sweet, J.; et al. Emergence of Enzalutamide resistance in prostate cancer is associated with BCL-2 and IKKB dependencies. Clin. Cancer Res. 2021, 27, 2340–2351. [Google Scholar] [CrossRef] [PubMed]
- Akfirat, C.; Zhang, X.; Ventura, A.; Berel, D.; Colangelo, M.E.; Miranti, C.K.; Krajewska, M.; Reed, J.C.; Higano, C.S.; True, L.D.; et al. Tumour cell survival mechanisms in lethal metastatic prostate cancer differ between bone and soft tissue metastases. J. Pathol. 2013, 230, 291–297. [Google Scholar] [CrossRef]
- Ali, A.; Kulik, G. Signaling pathways that control apoptosis in prostate cancer. Cancers 2021, 13, 937. [Google Scholar] [CrossRef]
- Santer, F.R.; Erb, H.H.; Oh, S.J.; Handle, F.; Feiersinger, G.E.; Luef, B.; Bu, H.; Schafer, G.; Ploner, C.; Egger, M.; et al. Mechanistic rationale for MCL1 inhibition during androgen deprivation therapy. Oncotarget 2015, 6, 6105–6122. [Google Scholar] [CrossRef] [Green Version]
- McDonnell, T.J.; Troncoso, P.; Brisbay, S.M.; Logothetis, C.; Chung, L.W.; Hsieh, J.T.; Tu, S.M.; Campbell, M.L. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 1992, 52, 6940–6944. [Google Scholar]
- Apakama, I.; Robinson, M.C.; Walter, N.M.; Charlton, R.G.; Royds, J.A.; Fuller, C.E.; Neal, D.E.; Hamdy, F.C. bcl-2 overexpression combined with p53 protein accumulation correlates with hormone-refractory prostate cancer. Br. J. Cancer 1996, 74, 1258–1262. [Google Scholar] [CrossRef]
- Li, Q.; Deng, Q.; Chao, H.P.; Liu, X.; Lu, Y.; Lin, K.; Liu, B.; Tang, G.W.; Zhang, D.; Tracz, A.; et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat. Commun. 2018, 9, 3600. [Google Scholar] [CrossRef] [Green Version]
- Kajiwara, T.; Takeuchi, T.; Ueki, T.; Moriyama, N.; Ueki, K.; Kakizoe, T.; Kawabe, K. Effect of Bcl-2 overexpression in human prostate cancer cells in vitro and in vivo. Int. J. Urol. 1999, 6, 520–525. [Google Scholar] [CrossRef]
- Huang, H.; Zegarra-Moro, O.L.; Benson, D.; Tindall, D.J. Androgens repress Bcl-2 expression via activation of the retinoblastoma (RB) protein in prostate cancer cells. Oncogene 2004, 23, 2161–2176. [Google Scholar] [CrossRef] [Green Version]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, J.; Alfaro, I.E.; Gomez, F.; Protter, A.A.; Bernales, S. Enzalutamide, an androgen receptor signaling inhibitor, induces tumor regression in a mouse model of castration-resistant prostate cancer. Prostate 2013, 73, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Pilling, A.B.; Hwang, C. Targeting prosurvival BCL2 signaling through Akt blockade sensitizes castration-resistant prostate cancer cells to enzalutamide. Prostate 2019, 79, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Corella, A.N.; Cabiliza Ordonio, M.V.A.; Coleman, I.; Lucas, J.M.; Kaipainen, A.; Nguyen, H.M.; Sondheim, D.; Brown, L.G.; True, L.D.; Lee, J.K.; et al. Identification of therapeutic vulnerabilities in small-cell neuroendocrine prostate cancer. Clin. Cancer Res. 2020, 26, 1667–1677. [Google Scholar] [CrossRef]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell 2017, 32, 474–489. [Google Scholar] [CrossRef] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachostergios, P.J.; Puca, L.; Beltran, H. Emerging variants of castration-resistant prostate cancer. Curr. Oncol. Rep. 2017, 19, 32. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [Green Version]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Harvey, C.T.; Geng, H.; Xue, C.; Chen, V.; Beer, T.M.; Qian, D.Z. Malate dehydrogenase 2 confers docetaxel resistance via regulations of JNK signaling and oxidative metabolism. Prostate 2013, 73, 1028–1037. [Google Scholar] [CrossRef] [Green Version]
- Karanika, S.; Karantanos, T.; Kurosaka, S.; Wang, J.; Hirayama, T.; Yang, G.; Park, S.; Golstov, A.A.; Tanimoto, R.; Li, L.; et al. GLIPR1-DeltaTM synergizes with docetaxel in cell death and suppresses resistance to docetaxel in prostate cancer cells. Mol. Cancer 2015, 14, 122. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.G.; Liu, X.M.; Kreis, W.; Budman, D.R. The effect of antimicrotubule agents on signal transduction pathways of apoptosis: A review. Cancer Chemother. Pharmacol. 1999, 44, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Haschka, M.D.; Karbon, G.; Soratroi, C.; O’Neill, K.L.; Luo, X.; Villunger, A. MARCH5-dependent degradation of MCL1/NOXA complexes defines susceptibility to antimitotic drug treatment. Cell Death Differ. 2020, 27, 2297–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz de Porras, V.; Wang, X.C.; Palomero, L.; Marin-Aguilera, M.; Sole-Blanch, C.; Indacochea, A.; Jimenez, N.; Bystrup, S.; Bakht, M.; Conteduca, V.; et al. Taxane-induced attenuation of the CXCR2/BCL-2 axis sensitizes prostate cancer to platinum-based treatment. Eur. Urol. 2021, 79, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Chintapalli, J.; Croce, C.M. Taxol induces bcl-2 phosphorylation and death of prostate cancer cells. Cancer Res. 1996, 56, 1253–1255. [Google Scholar]
- Mackey, T.J.; Borkowski, A.; Amin, P.; Jacobs, S.C.; Kyprianou, N. bcl-2/bax ratio as a predictive marker for therapeutic response to radiotherapy in patients with prostate cancer. Urology 1998, 52, 1085–1090. [Google Scholar] [CrossRef]
- Khor, L.Y.; Moughan, J.; Al-Saleem, T.; Hammond, E.H.; Venkatesan, V.; Rosenthal, S.A.; Ritter, M.A.; Sandler, H.M.; Hanks, G.E.; Shipley, W.U.; et al. Bcl-2 and Bax expression predict prostate cancer outcome in men treated with androgen deprivation and radiotherapy on radiation therapy oncology group protocol 92-02. Clin. Cancer Res. 2007, 13, 3585–3590. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Jonas, O.; Whitman, M.A.; Corey, E.; Balk, S.P.; Chen, S. Tyrosine kinase inhibitors increase MCL1 degradation and in combination with BCLXL/BCL2 inhibitors drive prostate cancer apoptosis. Clin. Cancer Res. 2018, 24, 5458–5470. [Google Scholar] [CrossRef] [Green Version]
- Leverson, J.D.; Zhang, H.; Chen, J.; Tahir, S.K.; Phillips, D.C.; Xue, J.; Nimmer, P.; Jin, S.; Smith, M.; Xiao, Y.; et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015, 6, e1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, N.; Skees, J.; Todd, K.J.; West, D.A.; Lambert, K.A.; Robinson, W.A.; Amato, C.M.; Couts, K.L.; Van Gulick, R.; MacBeth, M.; et al. MCL1 inhibitors S63845/MIK665 plus Navitoclax synergistically kill difficult-to-treat melanoma cells. Cell Death Dis. 2020, 11, 443. [Google Scholar] [CrossRef]
- Jackson, R.S., 2nd; Placzek, W.; Fernandez, A.; Ziaee, S.; Chu, C.Y.; Wei, J.; Stebbins, J.; Kitada, S.; Fritz, G.; Reed, J.C.; et al. Sabutoclax, a Mcl-1 antagonist, inhibits tumorigenesis in transgenic mouse and human xenograft models of prostate cancer. Neoplasia 2012, 14, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Stebbins, J.L.; Kitada, S.; Dash, R.; Zhai, D.; Placzek, W.J.; Wu, B.; Rega, M.F.; Zhang, Z.; Barile, E.; et al. An optically pure apogossypolone derivative as potent pan-active inhibitor of anti-apoptotic bcl-2 family proteins. Front. Oncol. 2011, 1, 28. [Google Scholar] [CrossRef] [Green Version]
- Sonpavde, G.; Matveev, V.; Burke, J.M.; Caton, J.R.; Fleming, M.T.; Hutson, T.E.; Galsky, M.D.; Berry, W.R.; Karlov, P.; Holmlund, J.T.; et al. Randomized phase II trial of docetaxel plus prednisone in combination with placebo or AT-101, an oral small molecule Bcl-2 family antagonist, as first-line therapy for metastatic castration-resistant prostate cancer. Ann. Oncol. 2012, 23, 1803–1808. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.N.; Hussain, M.; Stadler, W.M.; Liu, G.; Tereshchenko, I.V.; Goodin, S.; Jeyamohan, C.; Kaufman, H.L.; Mehnert, J.; DiPaola, R.S. A phase II study of AT-101 to overcome Bcl-2—Mediated resistance to androgen deprivation therapy in patients with newly diagnosed castration-sensitive metastatic prostate cancer. Clin. Genitourin. Cancer 2016, 14, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Sun, Y.; Huang, C.P.; You, B.; Ye, D.; Chang, C. Preclinical study using ABT263 to increase enzalutamide sensitivity to suppress prostate cancer progression via targeting BCL2/ROS/USP26 axis through altering ARv7 protein degradation. Cancers 2020, 12, 831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadal, R.; Schweizer, M.; Kryvenko, O.N.; Epstein, J.I.; Eisenberger, M.A. Small cell carcinoma of the prostate. Nat. Rev. Urol. 2014, 11, 213–219. [Google Scholar] [CrossRef]
- Tamaki, H.; Harashima, N.; Hiraki, M.; Arichi, N.; Nishimura, N.; Shiina, H.; Naora, K.; Harada, M. Bcl-2 family inhibition sensitizes human prostate cancer cells to docetaxel and promotes unexpected apoptosis under caspase-9 inhibition. Oncotarget 2014, 5, 11399–11412. [Google Scholar] [CrossRef] [Green Version]
- Parrondo, R.; de Las Pozas, A.; Reiner, T.; Perez-Stable, C. ABT-737, a small molecule Bcl-2/Bcl-xL antagonist, increases antimitotic-mediated apoptosis in human prostate cancer cells. PeerJ 2013, 1, e144. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Huang, S.B.; Yang, M.C.; Lin, Y.T.; Chu, I.H.; Shen, Y.N.; Chiu, Y.H.; Hung, S.H.; Kang, L.; Hong, Y.R.; et al. Combining paclitaxel with ABT-263 has a synergistic effect on paclitaxel resistant prostate cancer cells. PLoS ONE 2015, 10, e0120913. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Neuwirt, H.; Eder, I.E.; Kern, J.; Schafer, G.; Geley, S.; Heidegger, I.; Klocker, H.; Culig, Z. PIAS1 is a crucial factor for prostate cancer cell survival and a valid target in docetaxel resistant cells. Oncotarget 2014, 5, 12043–12056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Placzek, W.J. PTBP1 modulation of MCL1 expression regulates cellular apoptosis induced by antitubulin chemotherapeutics. Cell Death Differ. 2016, 23, 1681–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, K.S.R.; Smith, A.J.; Karpova, Y.; Datta, S.R.; Kulik, G. Diverse antiapoptotic signaling pathways activated by vasoactive intestinal polypeptide, epidermal growth factor, and phosphatidylinositol 3-kinase in prostate cancer cells converge on BAD. J. Biol. Chem. 2006, 281, 20891–20901. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.; Joshi, R.; Mathew, P. Synthetic lethality in PTEN-mutant prostate cancer is induced by combinatorial PI3K/Akt and BCL-XL inhibition. Mol. Cancer Res. 2016, 14, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Sastry, K.S.; Karpova, Y.; Kulik, G. Epidermal growth factor protects prostate cancer cells from apoptosis by inducing BAD phosphorylation via redundant signaling pathways. J. Biol. Chem. 2006, 281, 27367–27377. [Google Scholar] [CrossRef] [Green Version]
- Kinkade, C.W.; Castillo-Martin, M.; Puzio-Kuter, A.; Yan, J.; Foster, T.H.; Gao, H.; Sun, Y.; Ouyang, X.; Gerald, W.L.; Cordon-Cardo, C.; et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J. Clin. Invest. 2008, 118, 3051–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, C.R.; Balmanno, K.; Chalmers, C.; Hadfield, K.; Molton, S.A.; Ley, R.; Wagner, E.F.; Cook, S.J. Activation of ERK1/2 by deltaRaf-1:ER* represses Bim expression independently of the JNK or PI3K pathways. Oncogene 2003, 22, 1281–1293. [Google Scholar] [CrossRef] [Green Version]
- Nickols, N.G.; Nazarian, R.; Zhao, S.G.; Tan, V.; Uzunangelov, V.; Xia, Z.; Baertsch, R.; Neeman, E.; Gao, A.C.; Thomas, G.V.; et al. MEK-ERK signaling is a therapeutic target in metastatic castration resistant prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Gioeli, D.; Mandell, J.W.; Petroni, G.R.; Frierson, H.F., Jr.; Weber, M.J. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999, 59, 279–284. [Google Scholar] [PubMed]
- Arai, S.; Varkaris, A.; Nouri, M.; Chen, S.; Xie, L.; Balk, S.P. MARCH5 mediates NOXA-dependent MCL1 degradation driven by kinase inhibitors and integrated stress response activation. eLife 2020, 9, e54954. [Google Scholar] [CrossRef]
- Nakahara, T.; Kita, A.; Yamanaka, K.; Mori, M.; Amino, N.; Takeuchi, M.; Tominaga, F.; Hatakeyama, S.; Kinoyama, I.; Matsuhisa, A.; et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007, 67, 8014–8021. [Google Scholar] [CrossRef] [Green Version]
- Krajewska, M.; Krajewski, S.; Banares, S.; Huang, X.; Turner, B.; Bubendorf, L.; Kallioniemi, O.-P.; Shabaik, A.; Vitiello, A.; Peehl, D.; et al. Elevated expression of inhibitor of apoptosis proteins in prostate cancer. Clin. Cancer Res. 2003, 9, 4914–4925. [Google Scholar]

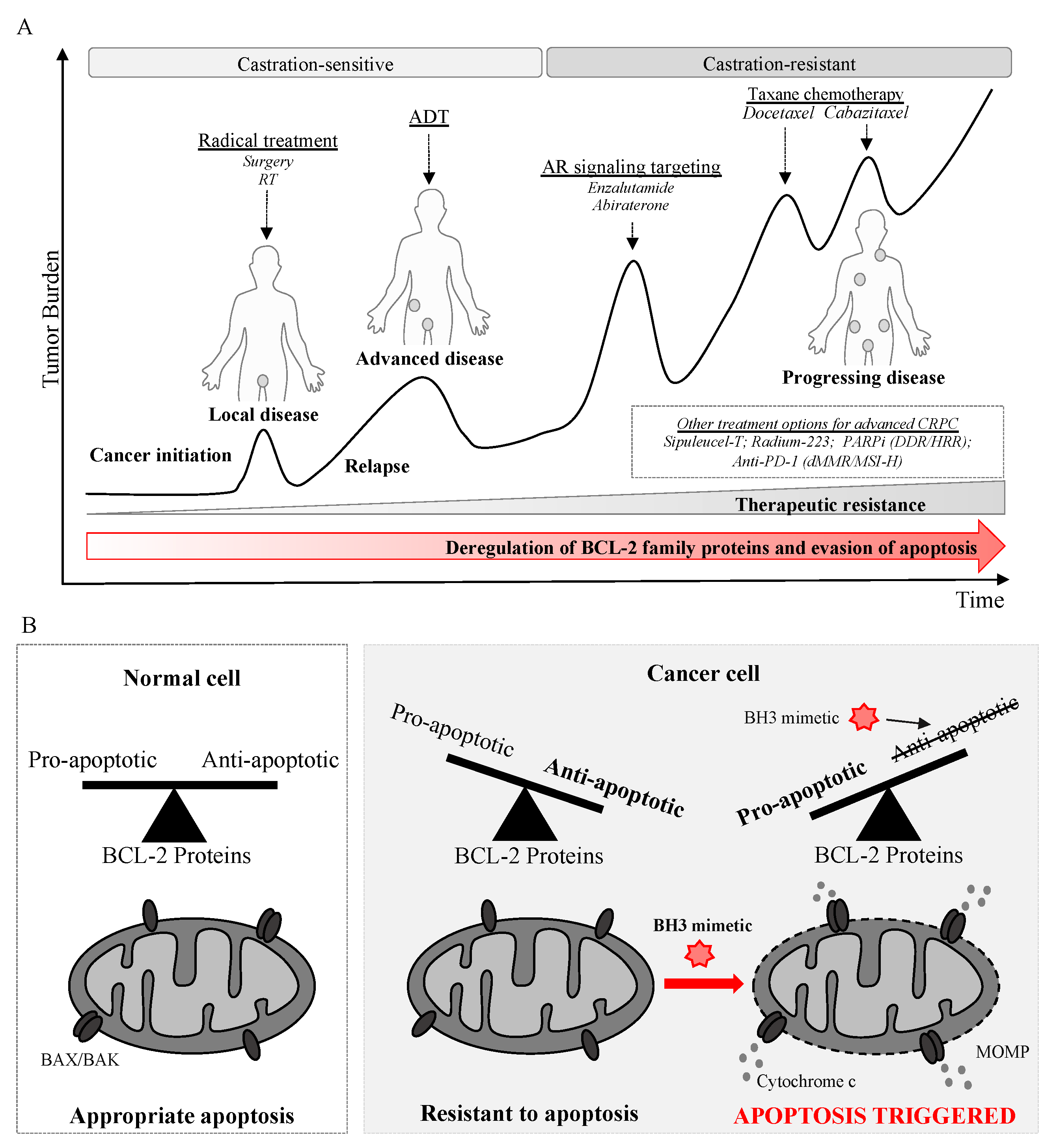

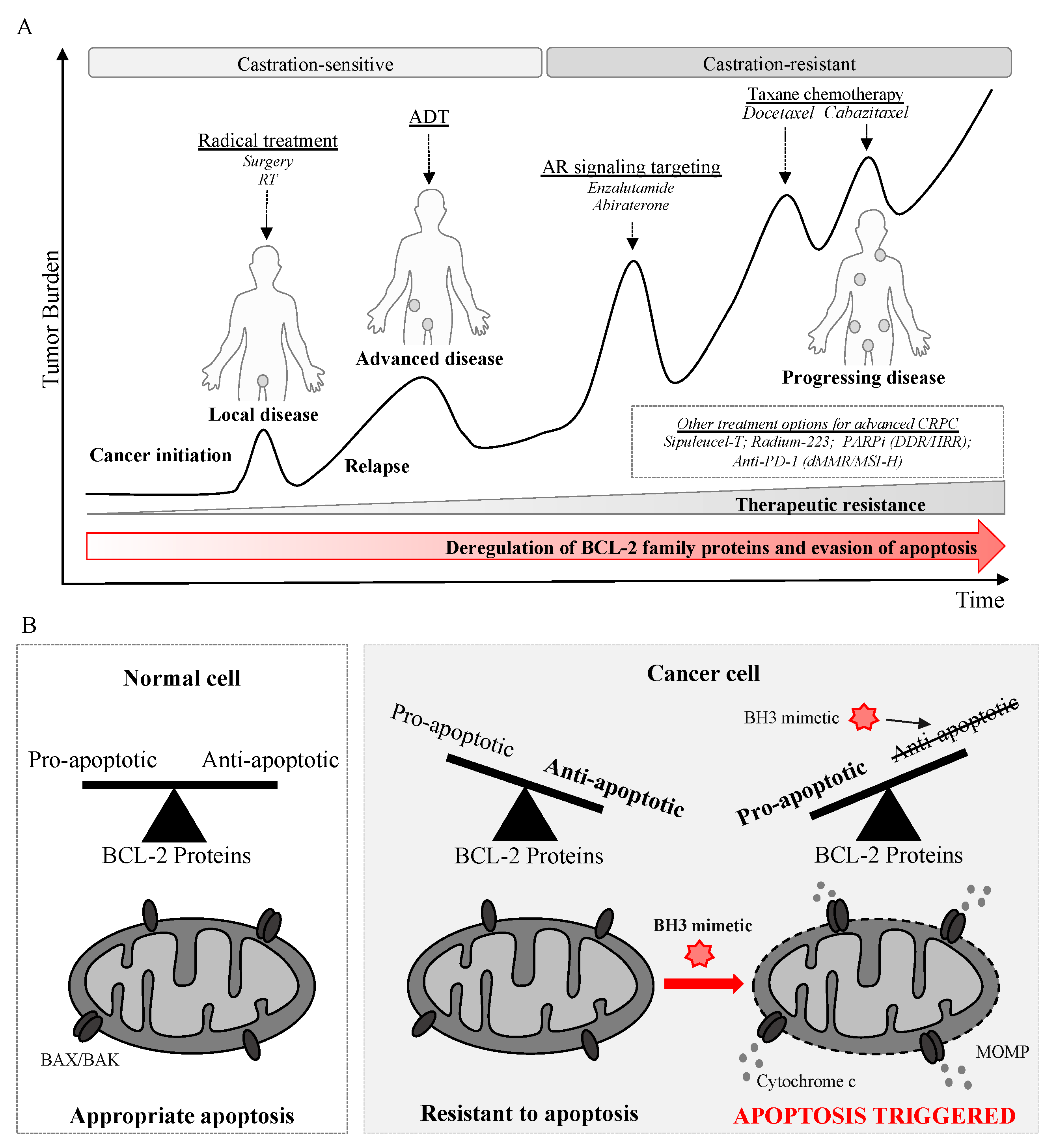
{kind=link}
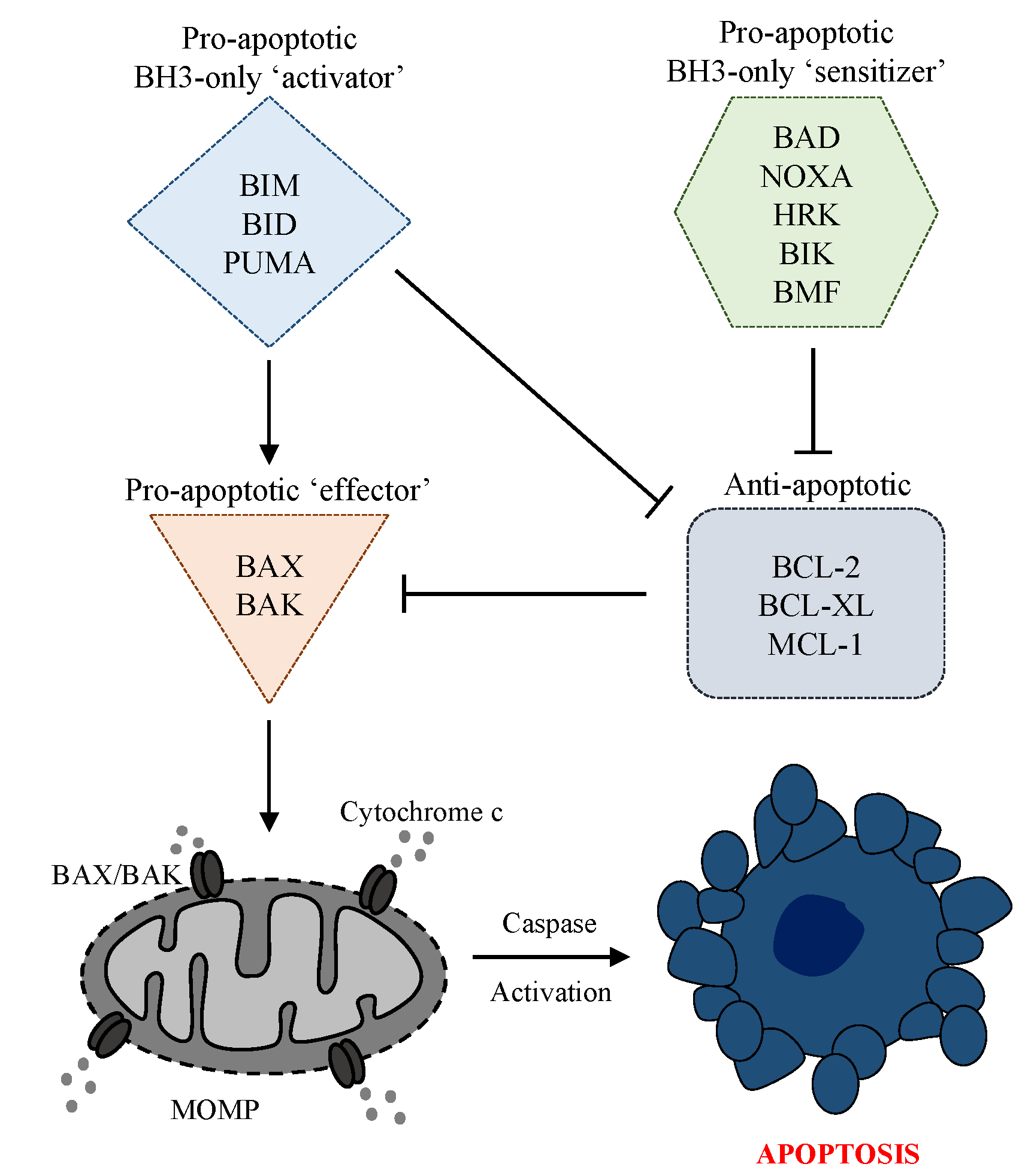
{kind=link}
| Morphotype | Stereotypical Morphological Changes | Diagram |
|---|---|---|
| Apoptosis (Type 1 cell death) |
|  |
| Autophagy (Type 2 cell death) |
| 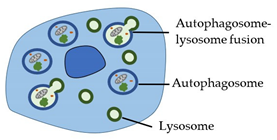 |
| Necrosis (Type 3 cell death) |
| 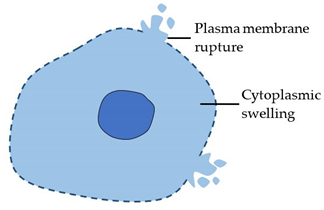 |
| Cell Death Type | Definition |
|---|---|
| Accidental cell death (ACD) | Rapid and uncontrolled cell death triggered by extreme physical, chemical, or mechanical insults and characterized by plasma membrane rupture. |
| Regulated cell death (RCD) | Highly coordinated cell death, dependent on the activation of one or more signal transduction pathways. RCD can be subdivided into numerous subroutines with significant interconnectivity, all of which can present with a range of morphological features (apoptotic to necrotic) and immunomodulatory effects. Subroutines include intrinsic apoptosis, extrinsic apoptosis, mitochondrial permeability transition (MPT)-driven necrosis, necroptosis, ferroptosis, pyroptosis, parthanatos, entotic cell death, NETotic cell death, lysosome-dependent cell death, autophagy-dependent cell death, and immunogenic cell death (see NCCD classification, Galluzzi et al.; 2018). |
| Programmed Cell Death (PCD) | Regulated cell death that occurs as part of normal physiological processes. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Westaby, D.; Jimenez-Vacas, J.M.; Padilha, A.; Varkaris, A.; Balk, S.P.; de Bono, J.S.; Sharp, A. Targeting the Intrinsic Apoptosis Pathway: A Window of Opportunity for Prostate Cancer. Cancers 2022, 14, 51. https://doi.org/10.3390/cancers14010051
Westaby D, Jimenez-Vacas JM, Padilha A, Varkaris A, Balk SP, de Bono JS, Sharp A. Targeting the Intrinsic Apoptosis Pathway: A Window of Opportunity for Prostate Cancer. Cancers. 2022; 14(1):51. https://doi.org/10.3390/cancers14010051
Chicago/Turabian StyleWestaby, Daniel, Juan M. Jimenez-Vacas, Ana Padilha, Andreas Varkaris, Steven P. Balk, Johann S. de Bono, and Adam Sharp. 2022. "Targeting the Intrinsic Apoptosis Pathway: A Window of Opportunity for Prostate Cancer" Cancers 14, no. 1: 51. https://doi.org/10.3390/cancers14010051
APA StyleWestaby, D., Jimenez-Vacas, J. M., Padilha, A., Varkaris, A., Balk, S. P., de Bono, J. S., & Sharp, A. (2022). Targeting the Intrinsic Apoptosis Pathway: A Window of Opportunity for Prostate Cancer. Cancers, 14(1), 51. https://doi.org/10.3390/cancers14010051