
HDAC Inhibition to Prime Immune Checkpoint Inhibitors



Abstract
:Simple Summary
Abstract
1. Introduction
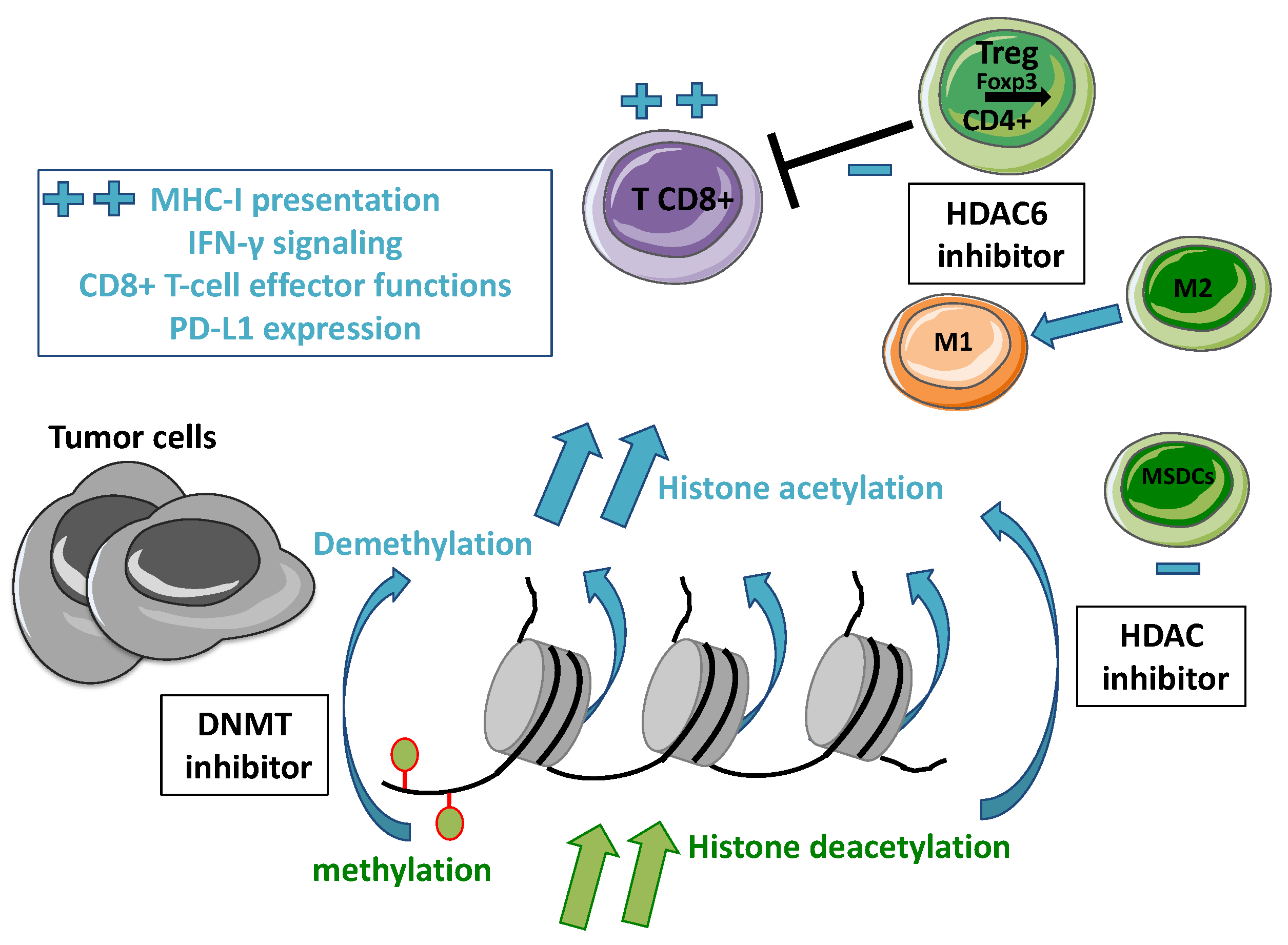
2. Overview of Epigenetic Alterations in Cancer
2.1. Epigenetic Regulations
2.2. DNA Methylation
2.3. Histone Modifications
3. Rationale for Combining an HDAC Inhibitor with Immunotherapy in Oncology
3.1. Immune Checkpoints Inhibition
3.2. Epigenetic Modulations to Restore Anti-Tumor Immune Response
3.3. Clinical Evidence of HDAC Inhibitors and ICI Combination
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. New Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswasi, S.; Allison, J.P. The Next Decade of Immune Checkpoint Therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Michalak, E.M.; Burr, M.L.; Bannister, A.J.; Dawson, M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Huttenhower, C.; Nosho, K.; Tanaka, N.; Shima, K.; Hazra, A.; Schernhammer, E.S.; Hunter, D.J.; Giovannucci, E.L.; Fuchs, C.S.; et al. Epigenomic diversity of colorectal cancer indicated by LINE-1 methylation in a database of 869 tumors. Mol. Cancer 2010, 9, 125. [Google Scholar] [CrossRef] [Green Version]
- Schübeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandze, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Lübbert, M.; Suciu, S.; Baila, L.; Rüter, B.H.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; Labar, B.; Germing, U.; Salih, H.R.; et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: Final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J. Clin. Oncol. 2011, 29, 1987–1996. [Google Scholar] [PubMed]
- Pleyer, L.; Greil, R. Digging deep into “dirty” drugs-modulation of the methylation machinery. Drug Metab. Rev. 2015, 47, 252–279. [Google Scholar] [CrossRef] [PubMed]
- Goutas, D.; Theocharis, S.; Tsourouflis, G. Unraveling the Epigenetic Role and Clinical Impact of Histone Deacetylases in Neoplasia. Diagnostics 2021, 11, 1346. [Google Scholar] [CrossRef] [PubMed]
- Martire, S.; Banaszynski, L.A. The roles of histone variants in fine-tuning chromatin organization and function. Nat. Rev. Mol. Cell Biol. 2020, 21, 522–541. [Google Scholar] [CrossRef] [PubMed]
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, L.F.; Zhang, D.-E. The 8;21 translocation in leukemogenesis. Oncogene 2004, 23, 4255–4262. [Google Scholar] [CrossRef] [Green Version]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016, 352, aad9780. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Grenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.; Doorduijn, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Wigle, T.J.; Warholic, N.M.; Sneeringer, C.J.; Allain, C.J.; Klaus, C.R.; Sacks, J.D.; Raimondi, A.; Majer, C.R.; Song, J.; et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012, 8, 890–896. [Google Scholar] [CrossRef]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenas-Ramirez, N.; Sahin, D.; Boyman, O. Epigenetic mechanisms of tumor resistance to immunotherapy. Cell Mol. Life Sci. 2018, 75, 4163–4176. [Google Scholar] [CrossRef]
- Ritter, C.; Fan, K.; Paschen, A.; Reker Hardrup, S.; Ferrone, S.; Nghiem, P.; Ugurel, S.; Schrama, D.; Becher, J.C. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep. 2017, 7, 2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.-W.C.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014, 5, 587–598. [Google Scholar]
- Adair, S.J.; Hogan, K.T. Treatment of ovarian cancer cell lines with 5-aza-2’-deoxycytidine upregulates the expression of cancer-testis antigens and class I major histocompatibility complex-encoded molecules. Cancer Immunol. Immunother. 2009, 58, 589–601. [Google Scholar] [CrossRef]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; Lafleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [Green Version]
- Sweet, M.J.; Shakespear, M.R.; Kamal, N.A.; Fairlie, D.P. HDAC inhibitors: Modulating leukocyte differentiation, survival, proliferation and inflammation. Immunol. Cell Biol. 2012, 90, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; DeStefano Shields, C.E.; Niknafs, N.; Yen, R.-W.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Van Criekinge, W.; De Meyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, R.; de Zoeten, E.F.; Ozkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 2007, 13, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Laino, A.S.; Betts, B.C.; Veerapathran, A.; Dolgalev, I.; Sarnaik, A.; Quayle, S.N.; Jones, S.S.; Weber, J.S.; Woods, D.M. HDAC6 selective inhibition of melanoma patient T-cells augments anti-tumor characteristics. J. Immunother. Cancer 2019, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Knox, T.; Sahakian, E.; Banik, D.; Hadley, M.; Palmer, E.; Noonepalle, S.; Kim, J.; Powers, J.; Gracia-Hernandez, M.; Oliveira, V.; et al. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci. Rep. 2019, 9, 6136. [Google Scholar] [CrossRef]
- Shen, S.; Hadley, M.; Ustinova, K.; Pavlicek, J.; Knox, T.; Noonepalle, S.; Tavares, M.T.; Zimprich, C.A.; Zhang, G.; Robers, M.B.; et al. Discovery of a New Isoxazole-3-hydroxamate-Based Histone Deacetylase 6 Inhibitor SS-208 with Antitumor Activity in Syngeneic Melanoma Mouse Models. J. Med. Chem. 2019, 62, 8557–8577. [Google Scholar] [CrossRef]
- Sahakian, E.; Powers, J.J.; Chen, J.; Deng, S.L.; Cheng, F.; Distler, A.; Woods, D.M.; Rock-Klotz, J.; Sodre, A.L.; Youn, J.-I.; et al. Histone deacetylase 11: A novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol. Immunol. 2015, 63, 579–585. [Google Scholar] [CrossRef] [Green Version]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [Green Version]
- Gray, J.E.; Saltos, A.; Tanvetyanon, T.; Haura, E.B.; Creelan, B.; Antonia, S.J.; Shafique, M.; Zheng, H.; Dai, W.; Saller, J.J.; et al. Phase I/Ib Study of Pembrolizumab Plus Vorinostat in Advanced/Metastatic Non-Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6623–6632. [Google Scholar] [CrossRef] [PubMed]
- Saltos, A.N.; Tanvetyanon, T.; Creelan, B.C.; Shafique, M.R.; Antonia, S.J.; Haura, E.B.; Zheng, H.; Barlow, L.; Saller, J.; Castellano-Fornelli, A.; et al. Phase II randomized trial of first-line pembrolizumab and vorinostat in patients with metastatic NSCLC (mNSCLC). J. Clin. Oncol. 2020, 38, 9567. [Google Scholar] [CrossRef]
- Rodriguez, C.P.; Wu, Q.; Voutsinas, J.; Fromm, J.R.; Jiang, X.; Pillarisetty, V.G.; Lee, S.M.; Santana-Davila, R.; Goulart, B.; Baik, C.S.; et al. A Phase II Trial of Pembrolizumab and Vorinostat in Recurrent Metastatic Head and Neck Squamous Cell Carcinomas and Salivary Gland Cancer. Clin. Cancer Res. 2020, 26, 837–845. [Google Scholar]
- Terranova-Barberio, M.; Pawlowska, N.; Dhawan, M.; Moasser, M.; Chien, A.J.; Melisko, M.E.; Rugo, H.; Rahimi, R.; Deal, T.; Daud, A.; et al. Exhausted T cell signature predicts immunotherapy response in ER-positive breast cancer. Nat. Commun. 2020, 11, 3584. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; Im, S.-A.; Lee, K.S.; Tan, A.; Telli, M.; Shachar, S.S.; Tchaleu, F.B.; Cha, E.; DuPree, B.; Nikanjam, M.; et al. 267P Phase Ib/II open-label, randomized evaluation of second- or third-line (2L/3L) atezolizumab (atezo) + entinostat (entino) in MORPHEUS-HR+ breast cancer (M-HR+BC). Ann. Oncol. 2021, 32, S479. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Moroose, R.L.; Babu, S.; Baramidze, K.; Chan, D.; Leitner, S.P.; Nemsadze, G.; Ordentilich, P.; Quaranto, C.; Meyers, M.L.; et al. Results of ENCORE 602 (TRIO025), a phase II, randomized, placebo-controlled, double-blinded, multicenter study of atezolizumab with or without entinostat in patients with advanced triple-negative breast cancer (aTNBC). J. Clin. Oncol. 2020, 38, 1014. [Google Scholar] [CrossRef]
- Pili, R.; Adra, N.; Damayanti, N.; Logan, T.F.; Narayan, V.; Monk, P.; Dropcho, S.; Sego, L.M.; Liu, H.; Althouse, S.K.; et al. Immunomodulation by HDAC inhibition: Results from a phase I study with entinostat in combination with atezolizumab and bevacizumab in metastatic renal cell carcinoma patients. J. Clin. Oncol. 2020, 38, 5064. [Google Scholar] [CrossRef]
- Roussos Torres, E.T.; Rafie, C.; Wang, C.; Lim, D.; Brufsky, A.; LoRusso, P.; Eder, J.P.; Chung, V.; Downs, M.; Geare, M.; et al. Phase I Study of Entinostat and Nivolumab with or without Ipilimumab in Advanced Solid Tumors (ETCTN-9844). Clin. Cancer Res. 2021, 27, 5828–5837. [Google Scholar] [CrossRef]
- Roussos Torres, E.T.; Leatherman, J.; Rafie, C.; Brufsky, A.; LoRusso, P.; Eder, J.P. Entinostat, nivolumab and ipilimumab in advanced HER2-negative breast cancer (ETCTN-9844). Ann. Oncol. 2021, 32, S829–S866. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Jänne, P.A.; Opyrchal, M.; Hafez, N.; Raez, L.E.; Gabrilovich, D.I.; Wang, F.; Trepel, J.B.; Lee, M.-J.; Yuno, A.; et al. Entinostat plus Pembrolizumab in Patients with Metastatic NSCLC Previously Treated with Anti-PD-(L)1 Therapy. Clin. Cancer Res. 2021, 27, 1019–1028. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Moschos, S.J.; Johnson, M.L.; Opyrchal, M.; Ordentlich, P.; Brouwer, S.; Sankoh, S.; Meyers, M.L.; Agarwala, S.S. Abstract CT072: Efficacy and safety of entinostat (ENT) and pembrolizumab (PEMBRO) in patients with melanoma previously treated with anti-PD1 therapy. Cancer Res. 2019, 79, CT072. [Google Scholar]
- Weber, J.S.; Laino, A.S.; Vassallo, M.; Pavlick, A.; Malatyali, S.; Krishnarajapet, S.; Deleon, G.; Chen, I.; Hallin, M.; Woods, D. Preclinical and clinical studies of a class I/IV HDAC inhibitor, mocetinostat, in melanoma. J. Clin. Oncol. 2020, 38, 10052. [Google Scholar] [CrossRef]
- Cartwright, E.; Turkes, F.; Saffery, C.; Tran, A.; Smith, G.; Esteban Moreno, S.; Hatt, S.; Renn, A.; Johnston, E.; Kohoutova, D.; et al. EMERGE: A phase II trial assessing the efficacy of domatinostat plus avelumab in patients with previously treated advanced mismatch repair proficient oesophagogastric and colorectal cancers–phase IIA dose finding. Ann. Oncol. 2021, 32, S555–S556. [Google Scholar] [CrossRef]
- Hassel, J.C.; Berking, C.; Schlaak, M.; Eigentler, T.; Gutzmer, R.; Ascierto, P.A.; Schilling, B.; Hamm, S.; Hermann, F.; Reimann, P.G.; et al. Results from the phase Ib of the SENSITIZE trial combining domatinostat with pembrolizumab in advanced melanoma patients refractory to prior checkpoint inhibitor therapy. J. Clin. Oncol. 2021, 39, 9545. [Google Scholar] [CrossRef]
- Sharma, P.; Abramson, V.G.; O’Dea, A.; Nye, L.E.; Mayer, I.A.; Crane, G.J.; Elia, M.; Yoder, R.; Staley, J.M.; Schwensen, K.; et al. Romidepsin (HDACi) plus cisplatin and nivolumab triplet combination in patients with metastatic triple negative breast cancer (mTNBC). J. Clin. Oncol. 2021, 39, 1076. [Google Scholar] [CrossRef]
- De Guillebon, E.; Jimenez, M.; Mazzarella, L.; Betsou, F.; Stadler, P.; Petak, I. Combining immunotherapy with an epidrug in squamous cell carcinomas of different locations: Rationale and design of the PEVO basket trial. ESMO Open 2021, 6, 100106. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.-G.; Yin, L.; Samsom, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Cai, S.; Wu, X.; Chen, Y.; Deng, H.; Zhong, X.; Chen, T.; Huang, M. A pan-cancer analysis of KMT2D as a potential biomarker for immune checkpoint therapy. Ann. Oncol. 2021, 32, S829–S866. [Google Scholar] [CrossRef]

{kind=link}
| HDAC Inhibitor | Immunotherapy Agents | Other Drugs | Cancer Type(s) | Trial Phase | Efficacy Results | NCT Number |
|---|---|---|---|---|---|---|
| Vorinostat | Pembrolizumab | NSCLC | I/IB | ORR = 13% [41] | 02638090 | |
| PD-L1 TPS ≥ 1% | II | ORR = 48% [42] | ||||
| Pembrolizumab | HNSCC, salivary gland tumors | II | HNSCC: ORR = 32% [43] | 02538510 | ||
| Pembrolizumab | Tamoxifen | HR positive breast cancer | II | ORR = 4% [44] CBR = 19% [44] | 02395627 | |
| Entinostat | Atezolizumab | HR positive breast cancer | I/II | ORR = 6.7% [45] | 03280563 | |
| Atezolizumab | TNBC | I/II | ORR = 10% [46] | 02708680 | ||
| CBR = 37.5% [46] | ||||||
| PFS = 1.68 mo. | ||||||
| Atezolizumab | Bevacizumab | RCC | I/II | ORR = 20% [47] | 03024437 | |
| PFS = 7.6 mo. | ||||||
| Nivolumab + ipilimumab | Advanced solid tumors | I | ORR = 16% [48] | 02453620 | ||
| HR+ and TNBC | II | ORR = 30% [49] | ||||
| Pembrolizumab | NSCLC with previous PD under ICI | II | ORR = 9.2% [50] | 02437136 | ||
| Pembrolizumab | Melanoma with previous PD under ICI | II | ORR = 19% [51] | 02437136 | ||
| Mocetinostat | Ipilimumab + Nivolumab | Melanoma | I | ORR = 70% [52] | 03565406 | |
| Domatinostat | Avelumab | Mismatch repair proficient CRC, oesophagogastric | IIA | SD = 46% [53] | 03812796 | |
| Pembrolizumab | Melanoma with previous PD under ICI | Ib | CBR= 30% [54] | 03278665 | ||
| Romidepsin | Nivolumab | Cisplatin | TN or BRCA-mutated breast cancer | I/II | ORR = 44% [55] | 02393794 |
| HDAC Inhibitor | Immunotherapy Agents | Other Drugs | Cancer Type(s) | Trial Phase | NCT Number |
|---|---|---|---|---|---|
| Vorinostat | Pembrolizumab | Renal or urothelial carcinoma | I/Ib | 02619253 | |
| Pembrolizumab | All types of SCC | II basket trial [56] | 04357873 | ||
| Entinostat | Pembrolizumab | Bladder cancer | II | 03978624 | |
| Pembrolizumab | Mismatch repair proficient CRC | II | 02437136 | ||
| Avelumab | Ovarian cancer Advanced | I/II | 02915523 | ||
| Bintrafusp Alpha + NHS-IL12 | solid tumors | I/II | 04708470 | ||
| HPV-refractory tumors | |||||
| Mocetinostat | Pembrolizumab | Guadecitabine | NSCLC | I/Ib | 03220477 |
| (DNMTi) | |||||
| Pembrolizumab | NSCLC | II | 02954991 | ||
| Durvalumab | Advanced solid tumor and NSCLC | I/II | 02805660 | ||
| Durvalumab | HNSCC | I | 02993991 | ||
| Chidamide | Toripalimab | Cervical cancer | I/II | 04651127 | |
| Nivolumab | NSCLC, RCC melanoma | I/II | 02718066 | ||
| Envafolimab | NSCLC with previous PD under ICI | II | 05068427 | ||
| Tirelizumab | Urothelial carcinoma | II | 04562311 | ||
| Panobinostat | Spartalizumab | NSCLC, CRC, TNBC | Ib | 02890069 | |
| Ipilimumab | Melanoma | I | 02032810 | ||
| Domatinostat | Nivolumab + Ipilimumab | Resectable muscle-invasive urothelial cancer | I | 04871594 | |
| Nivolumab + Ipilimumab | Stage III melanoma | I/II | 04133948 | ||
| Romidepsin | Pembrolizumab | Mismatch repair proficient CRC | I | 02512172 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borcoman, E.; Kamal, M.; Marret, G.; Dupain, C.; Castel-Ajgal, Z.; Le Tourneau, C. HDAC Inhibition to Prime Immune Checkpoint Inhibitors. Cancers 2022, 14, 66. https://doi.org/10.3390/cancers14010066
Borcoman E, Kamal M, Marret G, Dupain C, Castel-Ajgal Z, Le Tourneau C. HDAC Inhibition to Prime Immune Checkpoint Inhibitors. Cancers. 2022; 14(1):66. https://doi.org/10.3390/cancers14010066
Chicago/Turabian StyleBorcoman, Edith, Maud Kamal, Grégoire Marret, Celia Dupain, Zahra Castel-Ajgal, and Christophe Le Tourneau. 2022. "HDAC Inhibition to Prime Immune Checkpoint Inhibitors" Cancers 14, no. 1: 66. https://doi.org/10.3390/cancers14010066
APA StyleBorcoman, E., Kamal, M., Marret, G., Dupain, C., Castel-Ajgal, Z., & Le Tourneau, C. (2022). HDAC Inhibition to Prime Immune Checkpoint Inhibitors. Cancers, 14(1), 66. https://doi.org/10.3390/cancers14010066