
Physical Forces and Transient Nuclear Envelope Rupture during Metastasis: The Key for Success?



Abstract
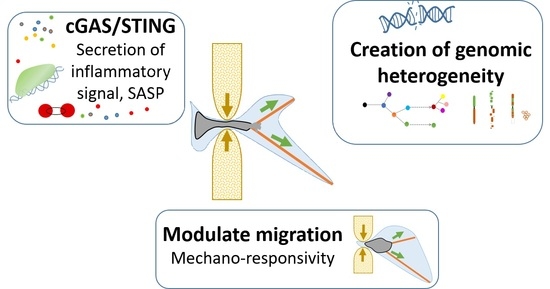
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Mechanical Stress during Metastasis
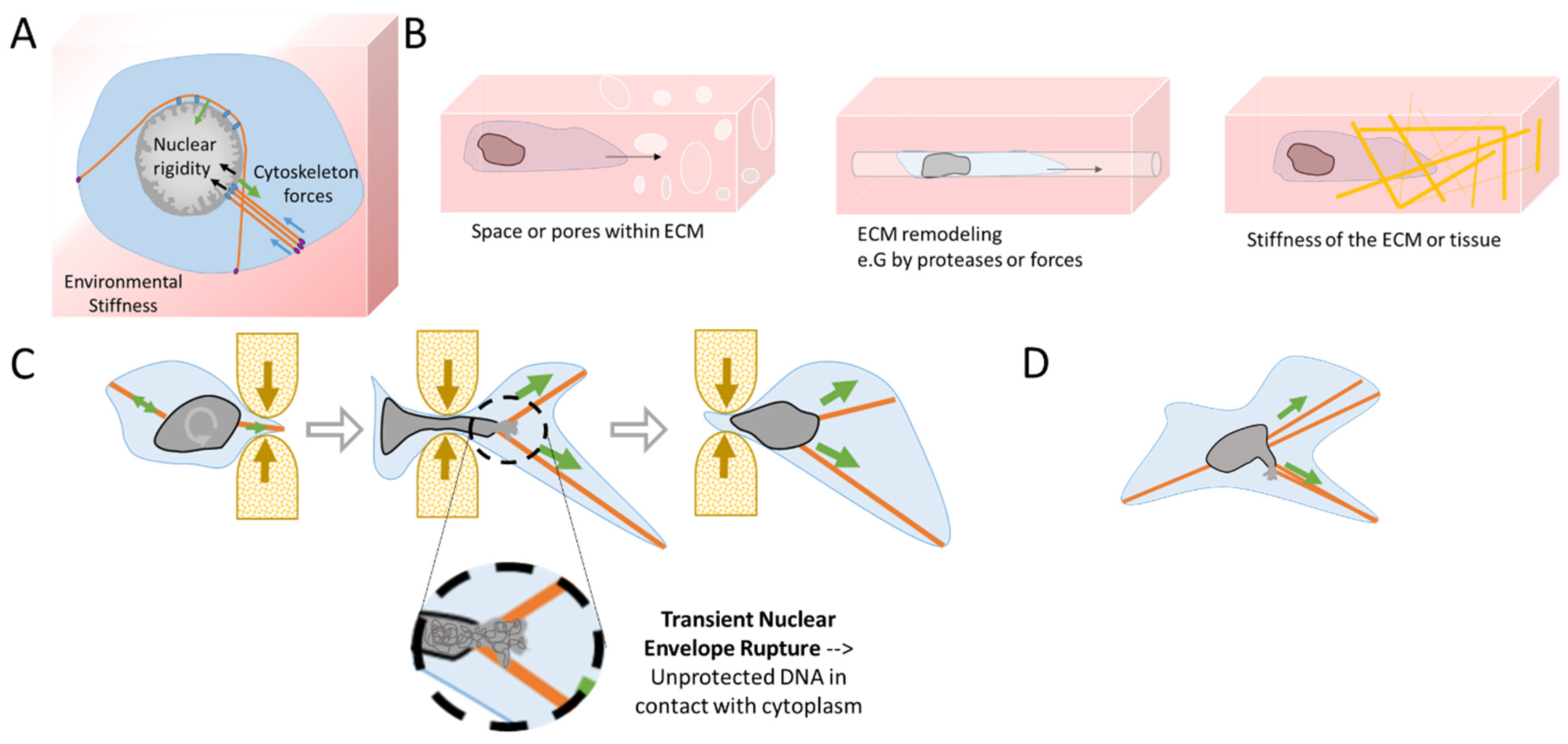
1.1. Mechanics behind Metastasis
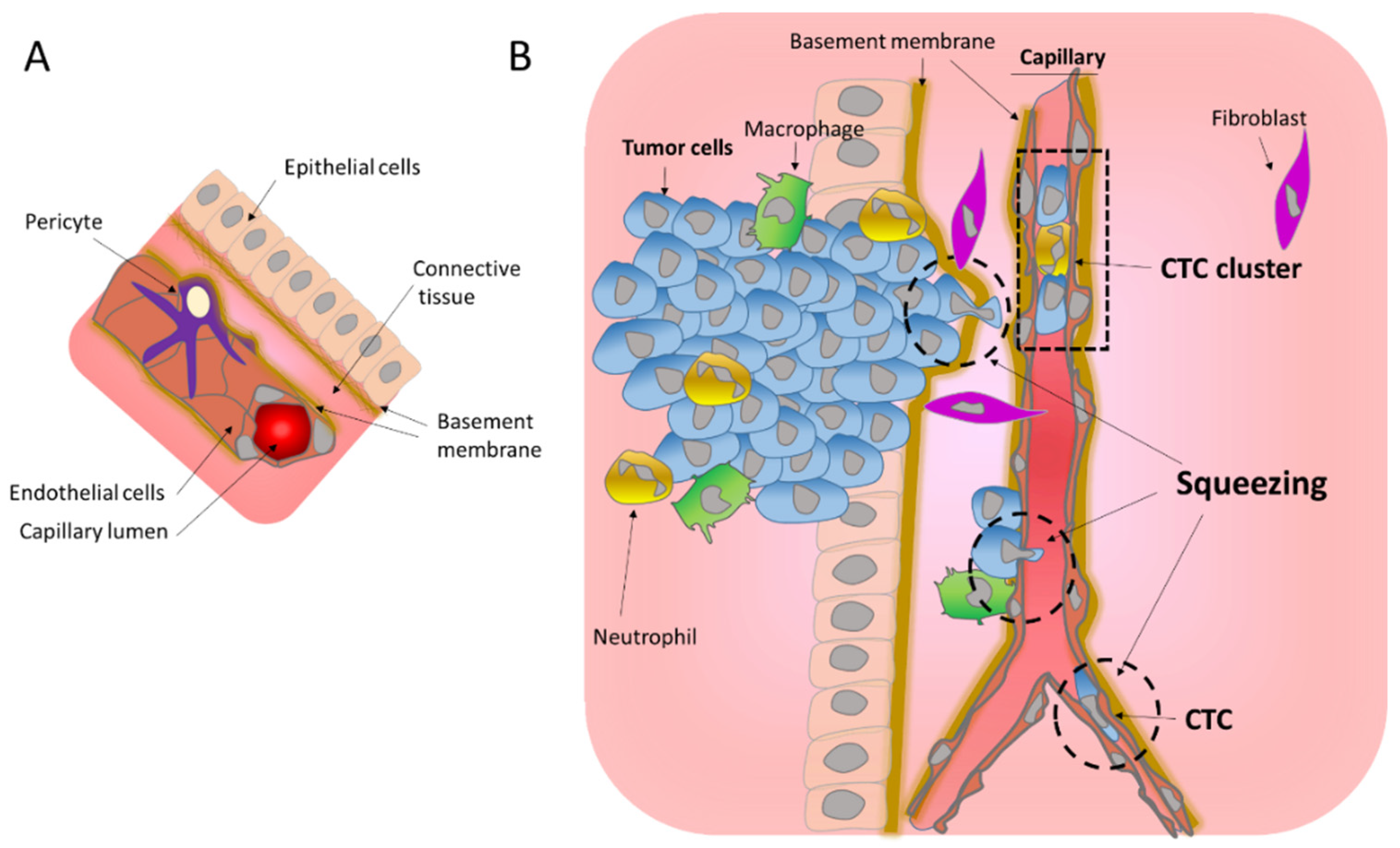
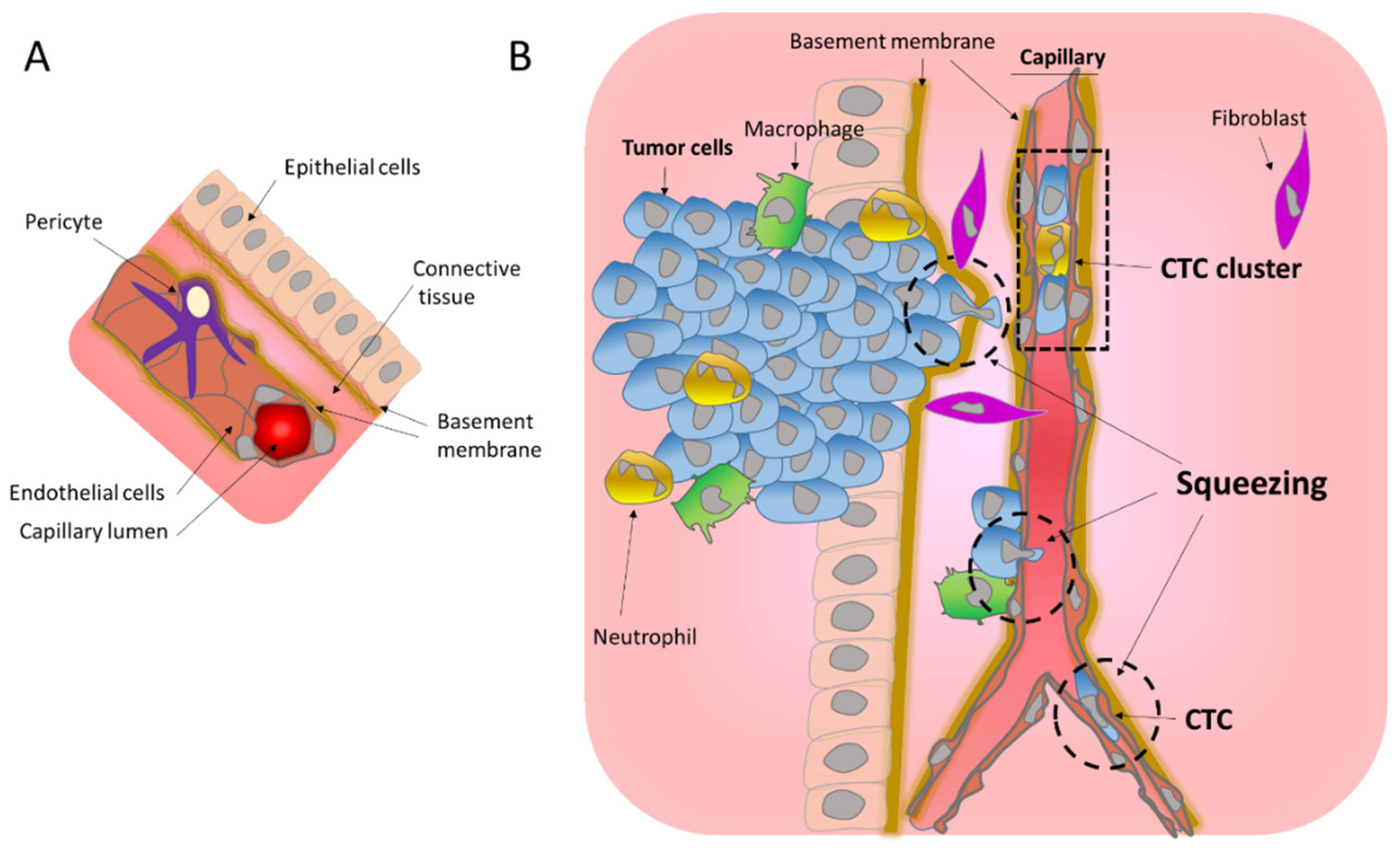
1.1.1. Migration through the Basement Membrane
1.1.2. Transendothelial Migration
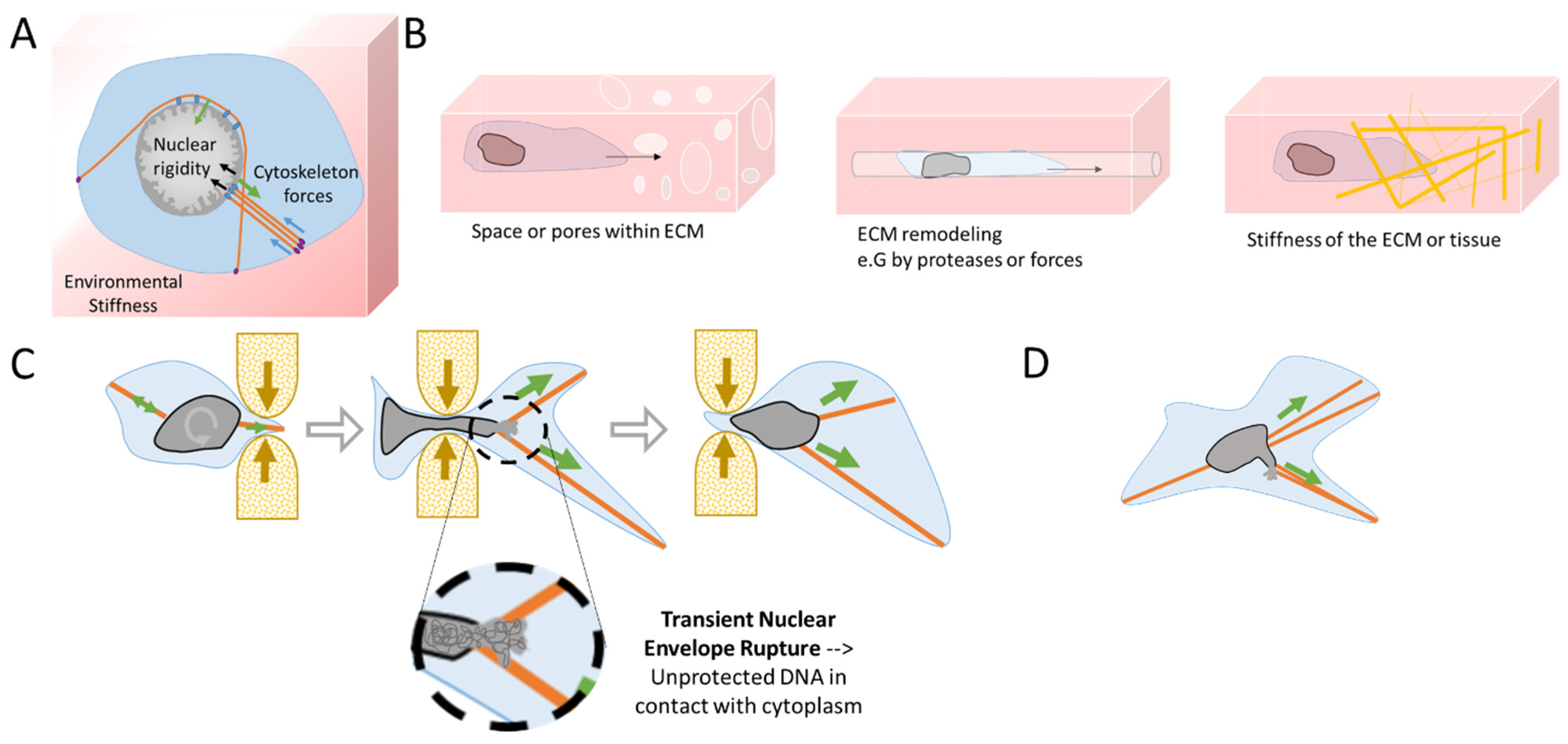
1.2. Cellular Plasticity and Mechanosensitivity under Migration
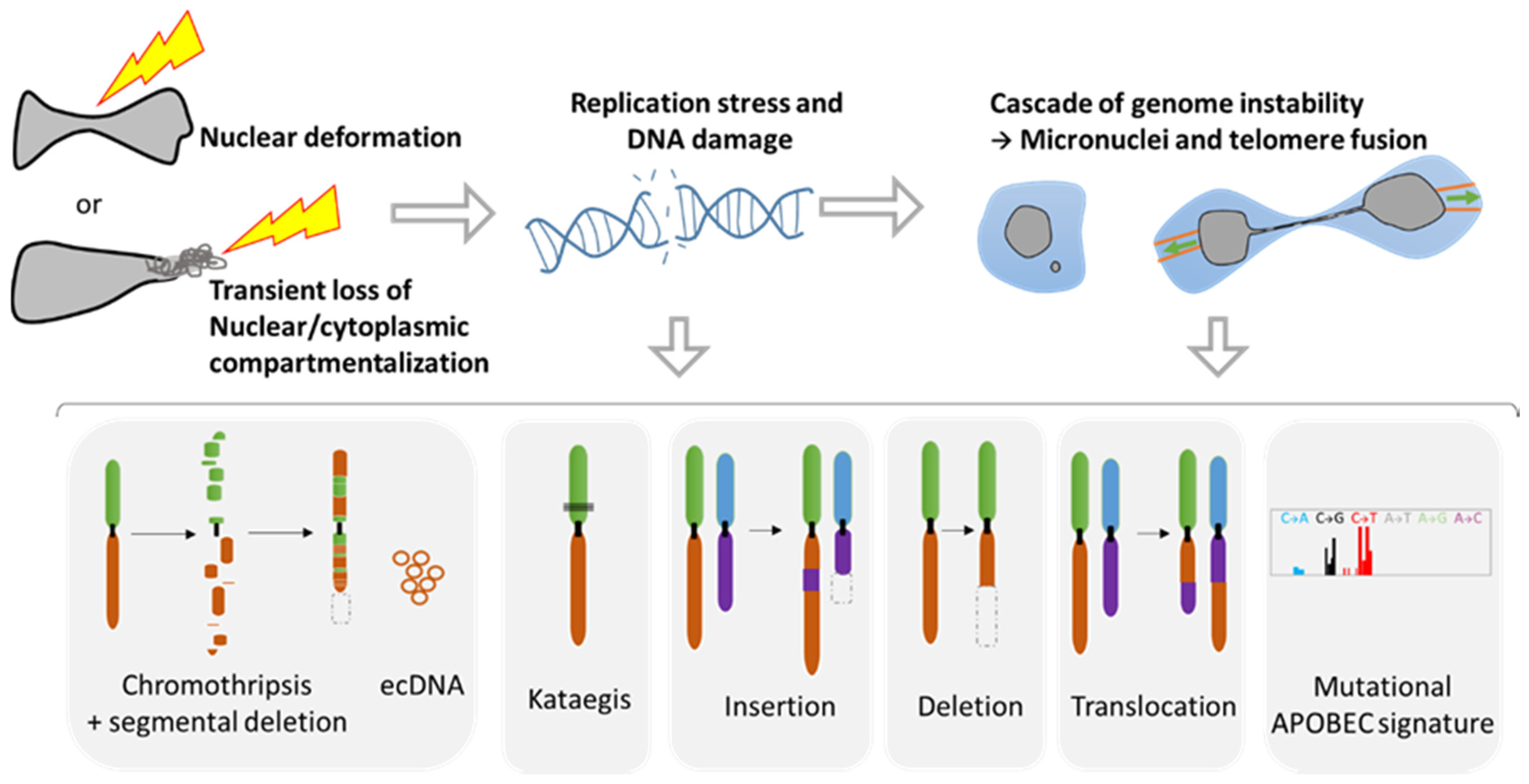
2. Consequences of Nuclear Envelope Rupture on Tumor Genomic Heterogeneity
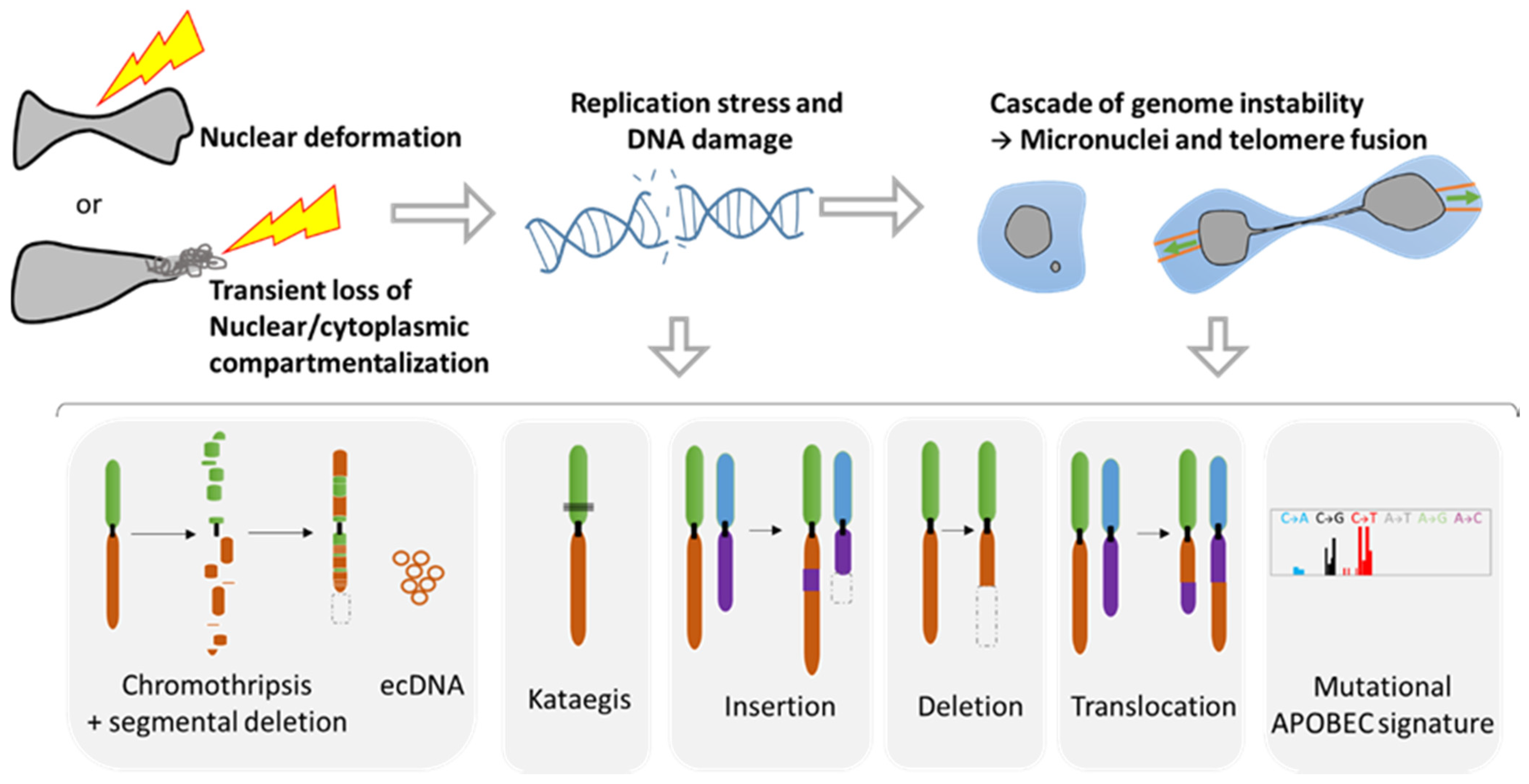
2.1. Role of NER in Simple and Complex Chromosomal Rearrangements
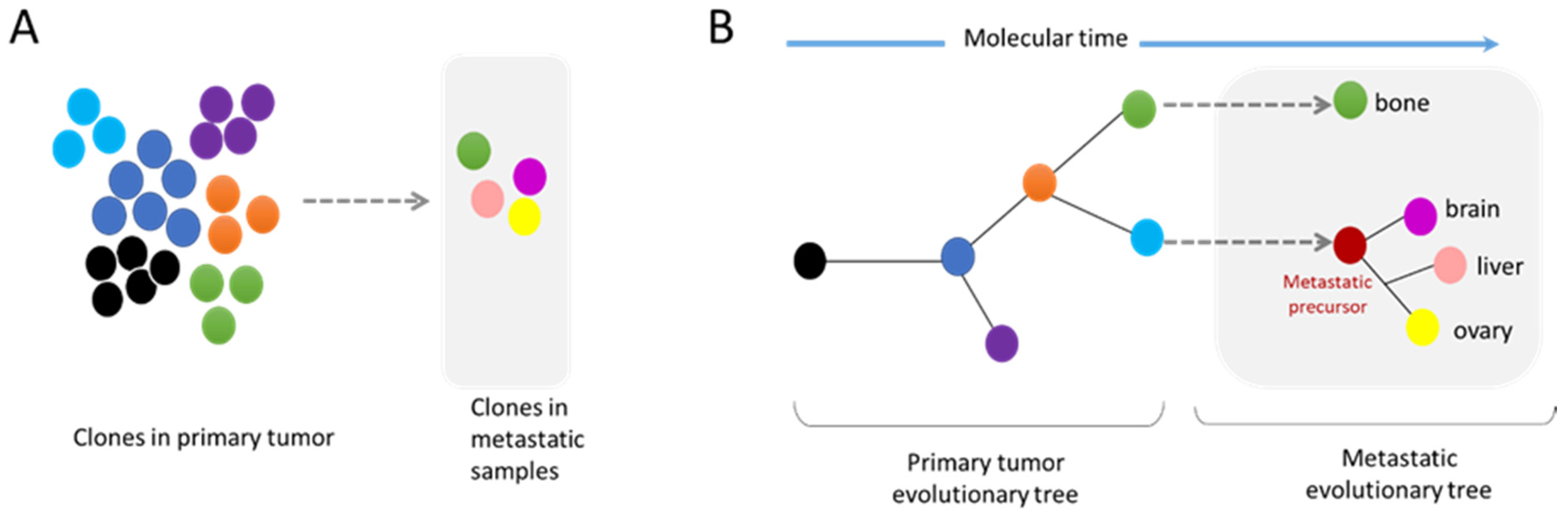
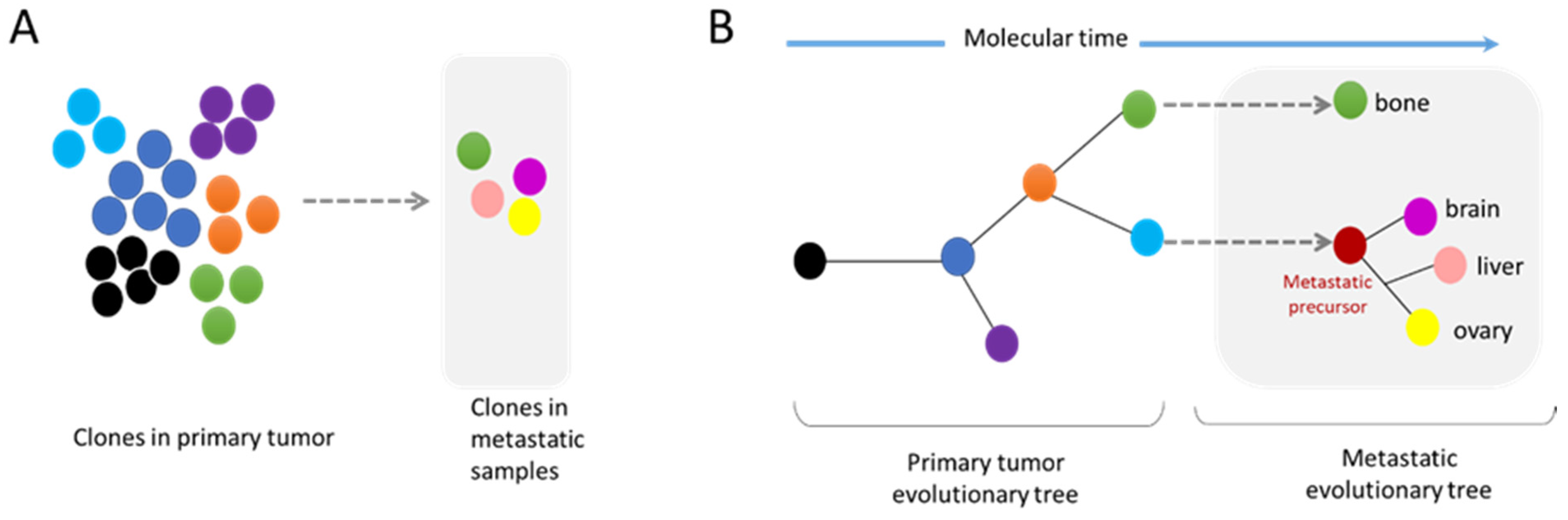
2.2. Metastasis and Genomic Evolution
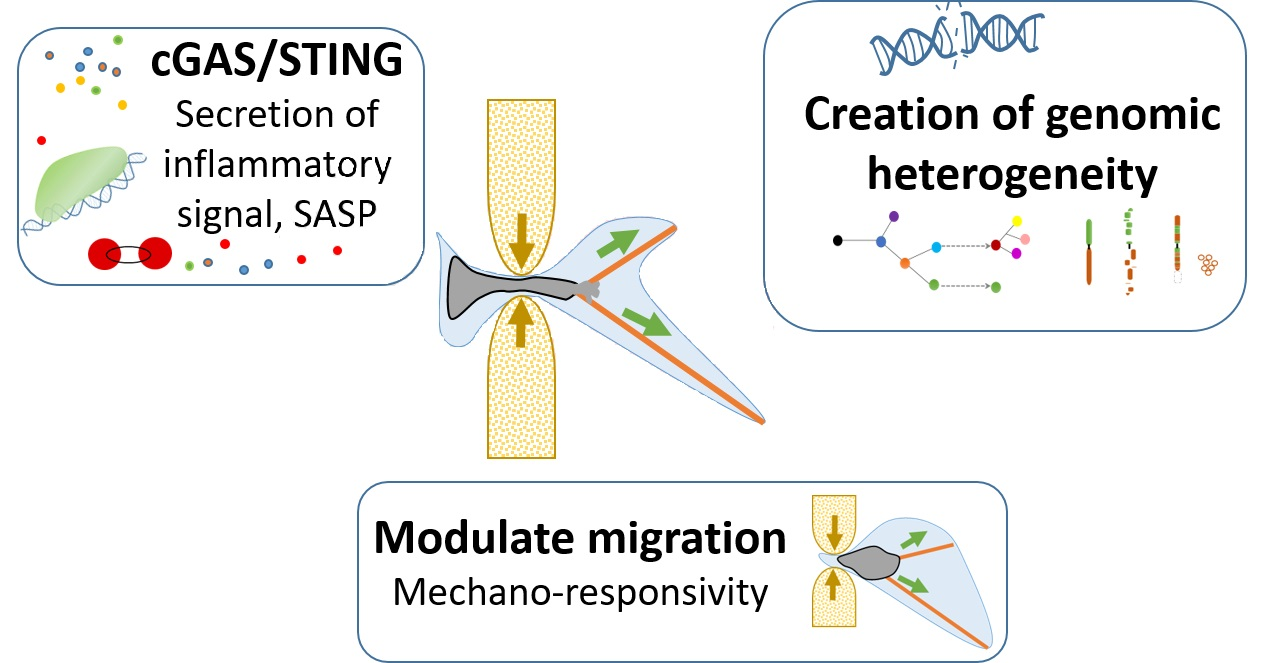
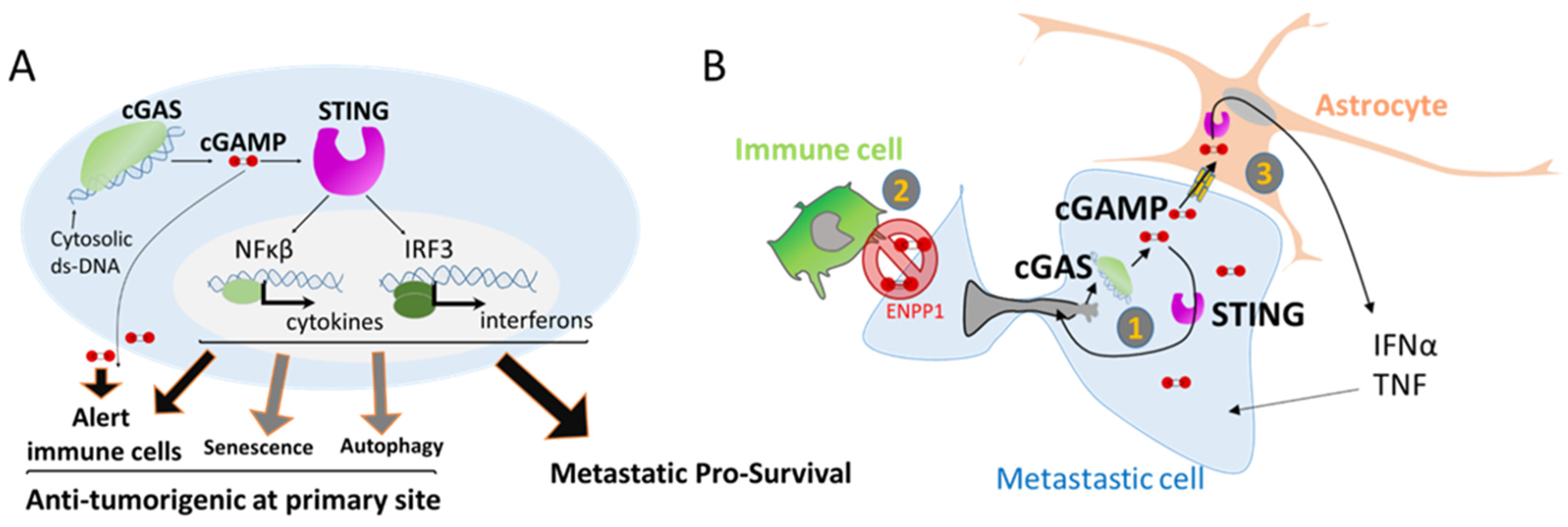
3. Nuclear Squeezing and Its Role in Activating the Innate Immune Response cGAS
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reuten, R.; Zendehroud, S.; Nicolau, M.; Fleischhauer, L.; Laitala, A.; Kiderlen, S.; Nikodemus, D.; Wullkopf, L.; Nielsen, S.R.; McNeilly, S.; et al. Basement membrane stiffness determines metastases formation. Nat. Mater. 2021, 20, 892–903. [Google Scholar] [CrossRef]
- Yurchenco, P.D. Basement membranes: Cell scaffoldings and signaling platforms. Cold Spring Harb. Perspect. Biol. 2011, 3, a004911. [Google Scholar] [CrossRef] [Green Version]
- Linder, S. Invadosomes at a glance. J. Cell Sci. 2009, 122, 3009–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, E.S.; Weaver, A.M. A new role for cortactin in invadopodia: Regulation of protease secretion. Eur. J. Cell Biol. 2008, 87, 581–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, C.; Di Martino, J.S.; Bravo-Cordero, J.J. Actin dynamics during tumor cell dissemination. Int. Rev. Cell Mol. Biol. 2021, 360, 65–98. [Google Scholar] [CrossRef] [PubMed]
- Sabeh, F.; Shimizu-Hirota, R.; Weiss, S.J. Protease-dependent versus -independent cancer cell invasion programs: Three-dimensional amoeboid movement revisited. J. Cell. Biol. 2009, 185, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Sznurkowska, M.K.; Aceto, N. The gate to metastasis: Key players in cancer cell intravasation. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.B.; Jones, J.G.; Condeelis, J.S.; Segall, J.E. A critical step in metastasis: In vivo analysis of intravasation at the primary tumor. Cancer Res. 2000, 60, 2504–2511. [Google Scholar]
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, K.M.; Adebowale, K.; Chang, J.; Lee, J.Y.; Nam, S.; Desai, R.; Rossen, N.S.; Rafat, M.; West, R.B.; Hodgson, L.; et al. Matrix mechanical plasticity regulates cancer cell migration through confining microenvironments. Nat. Commun. 2018, 9, 4144. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, P.; Martinez-Pena, I.; Pineiro, R. Dangerous Liaisons: Circulating Tumor Cells (CTCs) and Cancer-Associated Fibroblasts (CAFs). Cancers 2020, 12, 2861. [Google Scholar] [CrossRef] [PubMed]
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 2017, 8, 924. [Google Scholar] [CrossRef]
- Chang, J.; Chaudhuri, O. Beyond proteases: Basement membrane mechanics and cancer invasion. J. Cell. Biol. 2019, 218, 2456–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paavonsalo, S.; Hariharan, S.; Lackman, M.H.; Karaman, S. Capillary Rarefaction in Obesity and Metabolic Diseases-Organ-Specificity and Possible Mechanisms. Cells 2020, 9, 2683. [Google Scholar] [CrossRef] [PubMed]
- Barlow, K.D.; Sanders, A.M.; Soker, S.; Ergun, S.; Metheny-Barlow, L.J. Pericytes on the tumor vasculature: Jekyll or hyde? Cancer Microenviron. 2013, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Alon, R.; van Buul, J.D. Leukocyte Breaching of Endothelial Barriers: The Actin Link. Trends Immunol. 2017, 38, 606–615. [Google Scholar] [CrossRef]
- Gerhardt, T.; Ley, K. Monocyte trafficking across the vessel wall. Cardiovasc. Res. 2015, 107, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Maas, S.L.; Soehnlein, O.; Viola, J.R. Organ-Specific Mechanisms of Transendothelial Neutrophil Migration in the Lung, Liver, Kidney, and Aorta. Front. Immunol. 2018, 9, 2739. [Google Scholar] [CrossRef] [Green Version]
- Barzilai, S.; Yadav, S.K.; Morrell, S.; Roncato, F.; Klein, E.; Stoler-Barak, L.; Golani, O.; Feigelson, S.W.; Zemel, A.; Nourshargh, S.; et al. Leukocytes Breach Endothelial Barriers by Insertion of Nuclear Lobes and Disassembly of Endothelial Actin Filaments. Cell Rep. 2017, 18, 685–699. [Google Scholar] [CrossRef] [Green Version]
- Roh-Johnson, M.; Bravo-Cordero, J.J.; Patsialou, A.; Sharma, V.P.; Guo, P.; Liu, H.; Hodgson, L.; Condeelis, J. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene 2014, 33, 4203–4212. [Google Scholar] [CrossRef] [Green Version]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Iriondo, O.; Yu, M. Unexpected Friendship: Neutrophils Help Tumor Cells En Route to Metastasis. Dev. Cell 2019, 49, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Au, S.H.; Storey, B.D.; Moore, J.C.; Tang, Q.; Chen, Y.L.; Javaid, S.; Sarioglu, A.F.; Sullivan, R.; Madden, M.W.; O’Keefe, R.; et al. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc. Natl. Acad. Sci. USA 2016, 113, 4947–4952. [Google Scholar] [CrossRef] [Green Version]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Dogterom, M.; Koenderink, G.H. Actin-microtubule crosstalk in cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, B.R.; Comaills, V. Nuclear Envelope Integrity in Health and Disease: Consequences on Genome Instability and Inflammation. Int. J. Mol. Sci. 2021, 22, 7281. [Google Scholar] [CrossRef] [PubMed]
- Guilak, F.; Tedrow, J.R.; Burgkart, R. Viscoelastic properties of the cell nucleus. Biochem. Biophys. Res. Commun. 2000, 269, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Calero-Cuenca, F.J.; Janota, C.S.; Gomes, E.R. Dealing with the nucleus during cell migration. Curr. Opin. Cell Biol. 2018, 50, 35–41. [Google Scholar] [CrossRef]
- Maninova, M.; Iwanicki, M.P.; Vomastek, T. Emerging role for nuclear rotation and orientation in cell migration. Cell Adhes. Migr. 2014, 8, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.W.; Spinler, K.R.; Swift, J.; Chasis, J.A.; Mohandas, N.; Discher, D.E. Lamins regulate cell trafficking and lineage maturation of adult human hematopoietic cells. Proc. Natl. Acad. Sci. USA 2013, 110, 18892–18897. [Google Scholar] [CrossRef] [Green Version]
- Manley, H.R.; Keightley, M.C.; Lieschke, G.J. The Neutrophil Nucleus: An Important Influence on Neutrophil Migration and Function. Front. Immunol. 2018, 9, 2867. [Google Scholar] [CrossRef]
- Comaills, V.; Kabeche, L.; Morris, R.; Buisson, R.; Yu, M.; Madden, M.W.; LiCausi, J.A.; Boukhali, M.; Tajima, K.; Pan, S.; et al. Genomic Instability Is Induced by Persistent Proliferation of Cells Undergoing Epithelial-to-Mesenchymal Transition. Cell Rep. 2016, 17, 2632–2647. [Google Scholar] [CrossRef]
- Stroud, M.J.; Banerjee, I.; Veevers, J.; Chen, J. Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circ. Res. 2014, 114, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Wang, K.C.; Meng, Z. Mechanoregulation of YAP and TAZ in Cellular Homeostasis and Disease Progression. Front. Cell Dev. Biol. 2021, 9, 673599. [Google Scholar] [CrossRef]
- Rausch, V.; Hansen, C.G. The Hippo Pathway, YAP/TAZ, and the Plasma Membrane. Trends Cell Biol. 2020, 30, 32–48. [Google Scholar] [CrossRef] [PubMed]
- De Felice, D.; Alaimo, A. Mechanosensitive Piezo Channels in Cancer: Focus on altered Calcium Signaling in Cancer Cells and in Tumor Progression. Cancers 2020, 12, 1780. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Traynor, D.; Piel, M.; Kabla, A.J.; Kay, R.R. Pressure sensing through Piezo channels controls whether cells migrate with blebs or pseudopods. Proc. Natl. Acad. Sci. USA 2020, 117, 2506–2512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattet, L.; Jung, H.Y.; Matsumoto, M.W.; Aubol, B.E.; Kumar, A.; Adams, J.A.; Chen, A.C.; Sah, R.L.; Engler, A.J.; Pasquale, E.B.; et al. Matrix Rigidity Controls Epithelial-Mesenchymal Plasticity and Tumor Metastasis via a Mechanoresponsive EPHA2/LYN Complex. Dev. Cell 2020, 54, 302–316. [Google Scholar] [CrossRef]
- Yamada, K.M.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019, 20, 738–752. [Google Scholar] [CrossRef]
- Renkawitz, J.; Kopf, A.; Stopp, J.; de Vries, I.; Driscoll, M.K.; Merrin, J.; Hauschild, R.; Welf, E.S.; Danuser, G.; Fiolka, R.; et al. Nuclear positioning facilitates amoeboid migration along the path of least resistance. Nature 2019, 568, 546–550. [Google Scholar] [CrossRef]
- Nava, M.M.; Miroshnikova, Y.A.; Biggs, L.C.; Whitefield, D.B.; Metge, F.; Boucas, J.; Vihinen, H.; Jokitalo, E.; Li, X.; Garcia Arcos, J.M.; et al. Heterochromatin-Driven Nuclear Softening Protects the Genome against Mechanical Stress-Induced Damage. Cell 2020, 181, 800–817. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, A.J.; Cattin, C.J.; Cuvelier, D.; Alraies, Z.; Molina, M.; Nader, G.P.F.; Srivastava, N.; Saez, P.J.; Garcia-Arcos, J.M.; Zhitnyak, I.Y.; et al. The nucleus acts as a ruler tailoring cell responses to spatial constraints. Science 2020, 370, eaba2894. [Google Scholar] [CrossRef]
- Venturini, V.; Pezzano, F.; Catala Castro, F.; Hakkinen, H.M.; Jimenez-Delgado, S.; Colomer-Rosell, M.; Marro, M.; Tolosa-Ramon, Q.; Paz-Lopez, S.; Valverde, M.A.; et al. The nucleus measures shape changes for cellular proprioception to control dynamic cell behavior. Science 2020, 370, eaba2644. [Google Scholar] [CrossRef]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Dumenil, A.M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef]
- Vietri, M.; Schultz, S.W.; Bellanger, A.; Jones, C.M.; Petersen, L.I.; Raiborg, C.; Skarpen, E.; Pedurupillay, C.R.J.; Kjos, I.; Kip, E.; et al. Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat. Cell Biol. 2020, 22, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nader, G.P.F.; Aguera-Gonzalez, S.; Routet, F.; Gratia, M.; Maurin, M.; Cancila, V.; Cadart, C.; Palamidessi, A.; Ramos, R.N.; San Roman, M.; et al. Compromised nuclear envelope integrity drives TREX1-dependent DNA damage and tumor cell invasion. Cell 2021, 184, 5230–5246. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Hobson, C.M.; Cheng, S.; Colville, M.J.; Paszek, M.J.; Superfine, R.; Lammerding, J. Nuclear Deformation Causes DNA Damage by Increasing Replication Stress. Curr. Biol. 2021, 31, 753–765. [Google Scholar] [CrossRef]
- Pfeifer, C.R.; Xia, Y.; Zhu, K.; Liu, D.; Irianto, J.; Garcia, V.M.M.; Millan, L.M.S.; Niese, B.; Harding, S.; Deviri, D.; et al. Constricted migration increases DNA damage and independently represses cell cycle. Mol. Biol. Cell 2018, 29, 1948–1962. [Google Scholar] [CrossRef]
- Voronina, N.; Wong, J.K.L.; Hubschmann, D.; Hlevnjak, M.; Uhrig, S.; Heilig, C.E.; Horak, P.; Kreutzfeldt, S.; Mock, A.; Stenzinger, A.; et al. The landscape of chromothripsis across adult cancer types. Nat. Commun. 2020, 11, 2320. [Google Scholar] [CrossRef]
- Shoshani, O.; Brunner, S.F.; Yaeger, R.; Ly, P.; Nechemia-Arbely, Y.; Kim, D.H.; Fang, R.; Castillon, G.A.; Yu, M.; Li, J.S.Z.; et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2021, 591, 137–141. [Google Scholar] [CrossRef]
- Ly, P.; Brunner, S.F.; Shoshani, O.; Kim, D.H.; Lan, W.; Pyntikova, T.; Flanagan, A.M.; Behjati, S.; Page, D.C.; Campbell, P.J.; et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet. 2019, 51, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Umbreit, N.T.; Zhang, C.Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368, eaba0712. [Google Scholar] [CrossRef]
- Maciejowski, J.; Hatch, E.M. Nuclear Membrane Rupture and Its Consequences. Annu. Rev. Cell Dev. Biol. 2020, 36, 85–114. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [Green Version]
- Maciejowski, J.; Chatzipli, A.; Dananberg, A.; Chu, K.; Toufektchan, E.; Klimczak, L.J.; Gordenin, D.A.; Campbell, P.J.; de Lange, T. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat. Genet. 2020, 52, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Wang, C.; Ma, X.; Wang, E.; Peng, G. APOBEC3B, a molecular driver of mutagenesis in human cancers. Cell Biosci. 2017, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Gronroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef]
- Brown, D.; Smeets, D.; Szekely, B.; Larsimont, D.; Szasz, A.M.; Adnet, P.Y.; Rothe, F.; Rouas, G.; Nagy, Z.I.; Farago, Z.; et al. Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations. Nat. Commun. 2017, 8, 14944. [Google Scholar] [CrossRef] [Green Version]
- Birkbak, N.J.; McGranahan, N. Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2020, 37, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Klotz, R.; Thomas, A.; Teng, T.; Han, S.M.; Iriondo, O.; Li, L.; Restrepo-Vassalli, S.; Wang, A.; Izadian, N.; MacKay, M.; et al. Circulating Tumor Cells Exhibit Metastatic Tropism and Reveal Brain Metastasis Drivers. Cancer Discov. 2020, 10, 86–103. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef]
- Ragu, S.; Matos-Rodrigues, G.; Lopez, B.S. Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes 2020, 11, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 2019, 29, 1236–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Nassour, J.; Radford, R.; Correia, A.; Fuste, J.M.; Schoell, B.; Jauch, A.; Shaw, R.J.; Karlseder, J. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 2019, 565, 659–663. [Google Scholar] [CrossRef]
- Gonzalez-Meljem, J.M.; Apps, J.R.; Fraser, H.C.; Martinez-Barbera, J.P. Paracrine roles of cellular senescence in promoting tumourigenesis. Br. J. Cancer 2018, 118, 1283–1288. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P.; Salto-Tellez, M.; James, J.A.; et al. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov. 2021, 11, 1212–1227. [Google Scholar] [CrossRef]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A. 50+ shades of EMT in 20 years of embryo-cancer bonding. Nat. Rev. Mol. Cell Biol. 2020, 21, 563. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Mettenleiter, T.C. Breaching the Barrier-The Nuclear Envelope in Virus Infection. J. Mol. Biol. 2016, 428, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Marr, A.K.; Garcin, P.; Pante, N. Nuclear envelope disruption involving host caspases plays a role in the parvovirus replication cycle. J. Virol. 2011, 85, 4863–4874. [Google Scholar] [CrossRef] [Green Version]
- Fay, N.; Pante, N. Nuclear entry of DNA viruses. Front. Microbiol. 2015, 6, 467. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Behzad, A.R.; Carroll, J.B.; Pante, N. Parvoviral nuclear import: Bypassing the host nuclear-transport machinery. J. Gen. Virol. 2006, 87, 3209–3213. [Google Scholar] [CrossRef] [PubMed]
- Gasset-Rosa, F.; Chillon-Marinas, C.; Goginashvili, A.; Atwal, R.S.; Artates, J.W.; Tabet, R.; Wheeler, V.C.; Bang, A.G.; Cleveland, D.W.; Lagier-Tourenne, C. Polyglutamine-Expanded Huntingtin Exacerbates Age-Related Disruption of Nuclear Integrity and Nucleocytoplasmic Transport. Neuron 2017, 94, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, F.; Sousa, J.; Pereira, C.D.; da Cruz, E.S.O.A.B.; Rebelo, S. Nuclear envelope dysfunction and its contribution to the aging process. Aging Cell 2020, 19, e13143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, J.D.; Hatch, E.M.; Anderson, D.J.; Hetzer, M.W. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 2012, 3, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.Y.; Yang, Y.; Weston, T.A.; Belling, J.N.; Heizer, P.; Tu, Y.; Kim, P.; Edillo, L.; Jonas, S.J.; Weiss, P.S.; et al. An absence of lamin B1 in migrating neurons causes nuclear membrane ruptures and cell death. Proc. Natl. Acad. Sci. USA 2019, 116, 25870–25879. [Google Scholar] [CrossRef]
- De Vos, W.H.; Houben, F.; Kamps, M.; Malhas, A.; Verheyen, F.; Cox, J.; Manders, E.M.; Verstraeten, V.L.; van Steensel, M.A.; Marcelis, C.L.; et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum. Mol. Genet. 2011, 20, 4175–4186. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Giner, F.; Aceto, N. Tracking cancer progression: From circulating tumor cells to metastasis. Genome Med. 2020, 12, 31. [Google Scholar] [CrossRef] [Green Version]
- Mishima, Y.; Paiva, B.; Shi, J.; Park, J.; Manier, S.; Takagi, S.; Massoud, M.; Perilla-Glen, A.; Aljawai, Y.; Huynh, D.; et al. The Mutational Landscape of Circulating Tumor Cells in Multiple Myeloma. Cell Rep. 2017, 19, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Chemi, F.; Rothwell, D.G.; McGranahan, N.; Gulati, S.; Abbosh, C.; Pearce, S.P.; Zhou, C.; Wilson, G.A.; Jamal-Hanjani, M.; Birkbak, N.; et al. Pulmonary venous circulating tumor cell dissemination before tumor resection and disease relapse. Nat. Med. 2019, 25, 1534–1539. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, L.; Han, L.; Tuo, X.; Ma, S.; Wang, Y.; Feng, X.; Liang, D.; Sun, C.; Wang, Q.; et al. The Discordance of Gene Mutations between Circulating Tumor Cells and Primary/Metastatic Tumor. Mol. Ther. Oncolytics 2019, 15, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, C.; Cani, A.K.; Larios, J.M.; Hovelson, D.H.; Aung, K.; Darga, E.P.; Cannell, E.M.; Baratta, P.J.; Liu, C.J.; Chu, D.; et al. Comprehensive Mutation and Copy Number Profiling in Archived Circulating Breast Cancer Tumor Cells Documents Heterogeneous Resistance Mechanisms. Cancer Res. 2018, 78, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Keller, L.; Pantel, K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Rev. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Buisson, R.; Langenbucher, A.; Bowen, D.; Kwan, E.E.; Benes, C.H.; Zou, L.; Lawrence, M.S. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science 2019, 364, eaaw2872. [Google Scholar] [CrossRef]
- Langenbucher, A.; Bowen, D.; Sakhtemani, R.; Bournique, E.; Wise, J.F.; Zou, L.; Bhagwat, A.S.; Buisson, R.; Lawrence, M.S. An extended APOBEC3A mutation signature in cancer. Nat. Commun. 2021, 12, 1602. [Google Scholar] [CrossRef]
- Oh, S.; Bournique, E.; Bowen, D.; Jalili, P.; Sanchez, A.; Ward, I.; Dananberg, A.; Manjunath, L.; Tran, G.P.; Semler, B.L.; et al. Genotoxic stress and viral infection induce transient expression of APOBEC3A and pro-inflammatory genes through two distinct pathways. Nat. Commun. 2021, 12, 4917. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Comaills, V.; Burr, R.; Boulay, G.; Miyamoto, D.T.; Wittner, B.S.; Emmons, E.; Sil, S.; Koulopoulos, M.W.; Broderick, K.T.; et al. COX-2 mediates tumor-stromal prolactin signaling to initiate tumorigenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 5223–5232. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, C. Drug developers switch gears to inhibit STING. Nat. Biotechnol. 2019, 37, 199–201. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gauthier, B.R.; Lorenzo, P.I.; Comaills, V. Physical Forces and Transient Nuclear Envelope Rupture during Metastasis: The Key for Success? Cancers 2022, 14, 83. https://doi.org/10.3390/cancers14010083
Gauthier BR, Lorenzo PI, Comaills V. Physical Forces and Transient Nuclear Envelope Rupture during Metastasis: The Key for Success? Cancers. 2022; 14(1):83. https://doi.org/10.3390/cancers14010083
Chicago/Turabian StyleGauthier, Benoit R., Petra I. Lorenzo, and Valentine Comaills. 2022. "Physical Forces and Transient Nuclear Envelope Rupture during Metastasis: The Key for Success?" Cancers 14, no. 1: 83. https://doi.org/10.3390/cancers14010083
APA StyleGauthier, B. R., Lorenzo, P. I., & Comaills, V. (2022). Physical Forces and Transient Nuclear Envelope Rupture during Metastasis: The Key for Success? Cancers, 14(1), 83. https://doi.org/10.3390/cancers14010083