
Nelfinavir Induces Cytotoxicity towards High-Grade Serous Ovarian Cancer Cells, Involving Induction of the Unfolded Protein Response, Modulation of Protein Synthesis, DNA Damage, Lysosomal Impairment, and Potentiation of Toxicity Caused by Proteasome Inhibition

 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Cell Proliferation and Viability
2.3. Assessment of the Sensitivity of the PEO Cell Line Series to Cisplatin
2.4. Cell Cycle Analysis
2.5. Clonogenic Survival Assay following NFV Treatment
2.6. Measure of XBP1 mRNA Splicing
2.7. Western Blot Analysis
2.8. Puromycin Incorporation Assay
2.9. Autophagic Flux
2.10. Drug Interaction Analysis
2.11. Statistical Analysis
3. Results
3.1. Nelfinavir Inhibits Growth, Reduces Viability, Increases Hypo-Diploid DNA Content, and Blocks Clonogenic Survival of High-Grade Serous Ovarian Cancer (HGSOC) Cells Regardless of Platinum Sensitivity
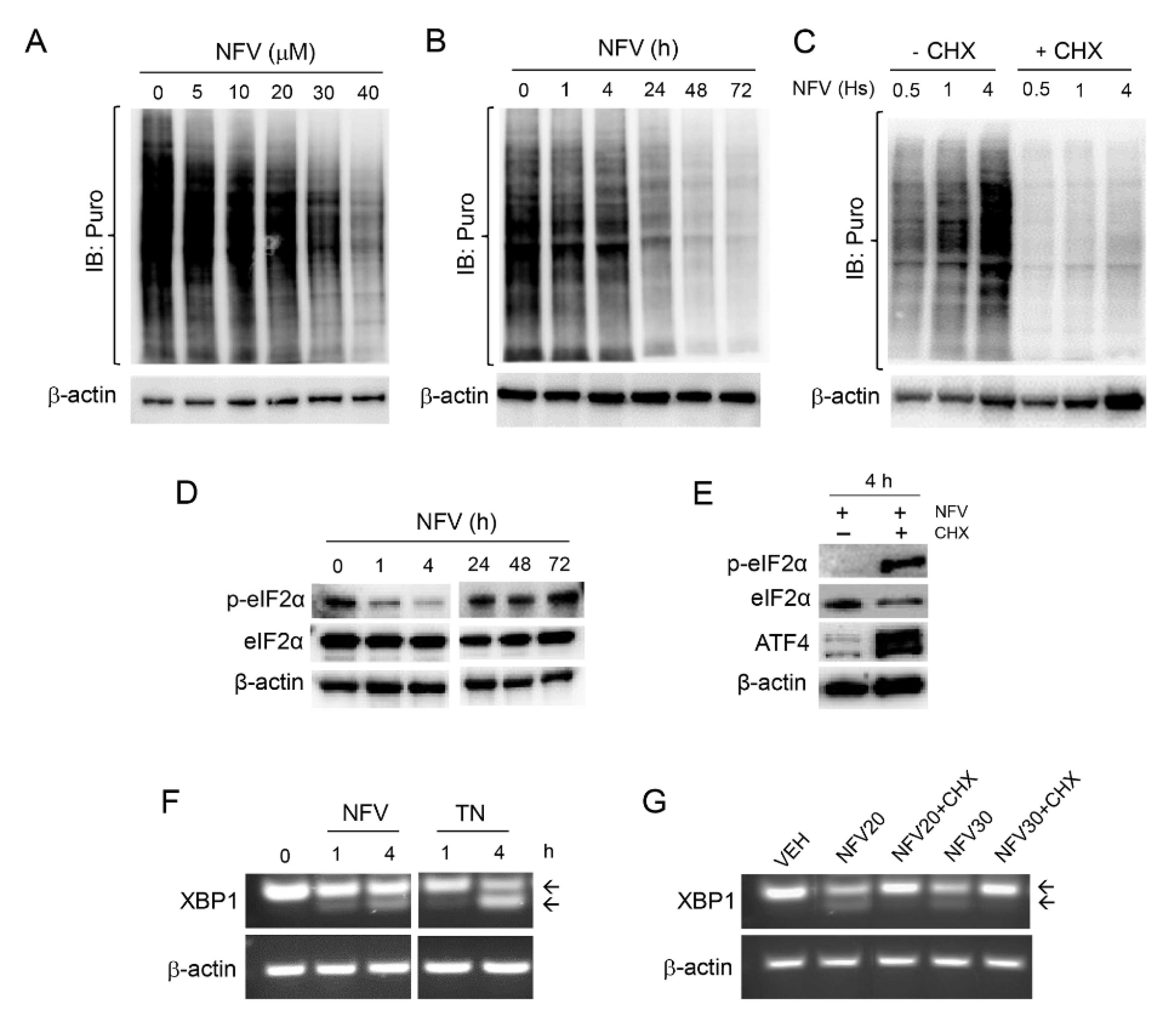
3.2. Nelfinavir Induces Cell Cycle Arrest, Triggers Endoplasmic Reticulum (ER) Stress and the Unfolded Protein Response (UPR), and Impairs the Function of Lysosomes without Affecting Autophagic Flux
3.3. ER Stress Response Induced by Nelfinavir Is Associated with Cleavage of Executioner Caspase-7 and Increased Pro-Apoptotic Bcl-2 Family Member Bax in a Time- and Concentration-Dependent Manner
3.4. Nelfinavir Toxicity Is Associated with Short-Term Sustained mRNA Translation That Contributes to the UPR, Followed by Long-Term Concentration-Dependent Inhibition of Global Protein Synthesis
3.5. Nelfinavir Inhibits AKT and ERK Phosphorylation and Triggers DNA Damage
3.6. Nelfinavir Potentiates the Toxicity of the Proteasome Inhibitor Bortezomib without Modifying Its Proteasome Inhibitory Capacity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDS | Acquired immunodeficiency syndrome |
| ANOVA | Analysis of variance |
| AKT | Protein kinase B |
| ATF4 | Activating transcription factor 4 |
| BAF | Bafilomycin A1 |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| BRCA | Breast cancer type 1 susceptibility protein |
| BZ | Bortezomib |
| CHOP | C/EBP homologous protein |
| CI | Combination index |
| Cdk-2 | Cyclin-dependent kinase 2 |
| CHX | Cycloheximide |
| CYP2C19 | Cytochrome P450 2C19 |
| DNA | Deoxyribonucleic acid |
| eIF2a | Eukaryotic initiation factor 2 alpha |
| ERAD | ER-associated degradation |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| GRP78 | Glucose-regulated protein, 78 kDa |
| HGSOC | High-grade serous ovarian cancer |
| HER2 | Receptor tyrosine-protein kinase erbB-2 |
| HIV | Human immunodeficiency viruses |
| HIV-PI | HIV protease inhibitors |
| HR | Homologous recombination |
| HSPA5 | Heat-shock 70 kDa protein 5 |
| H2AX | H2A histone family member X |
| IRE1a | Inositol-requiring enzyme 1α |
| KAP1 | KRAB-associated protein 1 |
| NFV | Nelfinavir |
| PARP | Poly (ADP-ribose) polymerase |
| PBS | Phosphate-buffered saline |
| PBMCs | Peripheral blood mononuclear cells |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| PFA | Paraformaldehyde |
| PI | Propidium iodide |
| PI3KCA | Constitutive active PI3K |
| P27kip1 | Cyclin dependent kinase inhibitor p27 (i.e., KIP1) |
| PTEN | Phosphatase and tensin homolog |
| RNA | Ribonucleic acid |
| RIP | Regulated intramembrane proteolysis |
| RT-PCR | Reverse transcription polymerase chain reaction |
| siRNA | Small interfering RNA |
| sATF6 | Soluble transcription factor ATF6 |
| S2P | Site-2 protease |
| STR | Autosomal short tandem repeat |
| TN | Tunicamycin |
| UPR | Unfolded protein response |
| XBP1 | X-box binding protein 1 |
References
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.; Matulonis, U. Next-Generation Sequencing: Role in Gynecologic Cancers. J. Natl. Compr. Canc. Netw. 2016, 14, 1165–1173. [Google Scholar] [CrossRef]
- Ota, S.; Ushijima, K.; Fujiyoshi, N.; Fujimoto, T.; Hayashi, R.; Murakami, F.; Komai, K.; Fujiyoshi, K.; Hori, D.; Kamura, T. Desmoplastic small round cell tumor in the ovary: Report of two cases and literature review. J. Obstet. Gynaecol. Res. 2010, 36, 430–434. [Google Scholar] [CrossRef]
- Ushijima, K. Treatment for recurrent ovarian cancer-at first relapse. J. Oncol. 2010, 2010, 497429. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [PubMed]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Flexner, C. HIV-protease inhibitors. N. Engl. J. Med. 1998, 338, 1281–1292. [Google Scholar] [CrossRef]
- Pai, V.B.; Nahata, M.C. Nelfinavir mesylate: A protease inhibitor. Ann. Pharmacother. 1999, 33, 325–339. [Google Scholar] [CrossRef]
- Subeha, M.R.; Telleria, C.M. The Anti-Cancer Properties of the HIV Protease Inhibitor Nelfinavir. Cancers 2020, 12, 3437. [Google Scholar] [CrossRef]
- Gills, J.J.; Lopiccolo, J.; Tsurutani, J.; Shoemaker, R.H.; Best, C.J.; Abu-Asab, M.S.; Borojerdi, J.; Warfel, N.A.; Gardner, E.R.; Danish, M.; et al. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin. Cancer Res. 2007, 13, 5183–5194. [Google Scholar] [CrossRef]
- Shim, J.S.; Rao, R.; Beebe, K.; Neckers, L.; Han, I.; Nahta, R.; Liu, J.O. Selective inhibition of HER2-positive breast cancer cells by the HIV protease inhibitor nelfinavir. J. Natl. Cancer Inst. 2012, 104, 1576–1590. [Google Scholar] [CrossRef]
- Mathur, A.; Elmageed, Z.Y.A.; Liu, X.; Kostochka, M.L.; Zhang, H.; Abdel-Mageed, A.B.; Mondal, D. Subverting ER-stress towards apoptosis by nelfinavir and curcumin coexposure augments docetaxel efficacy in castration resistant prostate cancer cells. PLoS ONE 2014, 9, e103109. [Google Scholar] [CrossRef] [PubMed]
- Bono, C.; Karlin, L.; Harel, S.; Mouly, E.; Labaume, S.; Galicier, L.; Apcher, S.; Sauvageon, H.; Fermand, J.P.; Bories, J.C.; et al. The human immunodeficiency virus-1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the proliferation of multiple myeloma cells in vitro and in vivo. Haematologica 2012, 97, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Bruning, A.; Rahmeh, M.; Gingelmaier, A.; Friese, K. The mitochondria-independent cytotoxic effect of nelfinavir on leukemia cells can be enhanced by sorafenib-mediated mcl-1 downregulation and mitochondrial membrane destabilization. Mol. Cancer 2010, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Mikochik, P.J.; Ra, J.H.; Lei, H.; Flaherty, K.T.; Winkler, J.D.; Spitz, F.R. HIV protease inhibitor nelfinavir inhibits growth of human melanoma cells by induction of cell cycle arrest. Cancer Res. 2007, 67, 1221–1227. [Google Scholar] [CrossRef]
- Bruning, A.; Burger, P.; Vogel, M.; Rahmeh, M.; Gingelmaiers, A.; Friese, K.; Lenhard, M.; Burges, A. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol. Ther. 2009, 8, 226–232. [Google Scholar] [CrossRef]
- Langdon, S.P.; Lawrie, S.S.; Hay, F.G.; Hawkes, M.M.; McDonald, A.; Hayward, I.P.; Schol, D.J.; Hilgers, J.; Leonard, R.C.; Smyth, J.F. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 1988, 48, 6166–6172. [Google Scholar]
- Cooke, S.L.; Ng, C.K.; Melnyk, N.; Garcia, M.J.; Hardcastle, T.; Temple, J.; Langdon, S.; Huntsman, D.; Brenton, J.D. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene 2010, 29, 4905–4913. [Google Scholar] [CrossRef]
- Goyeneche, A.; Lisio, M.A.; Fu, L.; Srinivasan, R.; Capuccino, J.V.; Gao, Z.H.; Telleria, C. The Capacity of High-Grade Serous Ovarian Cancer Cells to Form Multicellular Structures Spontaneously along Disease Progression Correlates with Their Orthotopic Tumorigenicity in Immunosuppressed Mice. Cancers 2020, 12, 699. [Google Scholar] [CrossRef]
- Freeburg, E.M.; Goyeneche, A.A.; Telleria, C.M. Mifepristone abrogates repopulation of ovarian cancer cells in between courses of cisplatin treatment. Int. J. Oncol. 2009, 34, 743–755. [Google Scholar] [CrossRef]
- Taylor, S.C.; Berkelman, T.; Yadav, G.; Hammond, M. A defined methodology for reliable quantification of Western blot data. Mol. Biotechnol. 2013, 55, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, Y.; Stoleru, D.; Salic, A. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc. Natl. Acad. Sci. USA 2012, 109, 413–418. [Google Scholar] [CrossRef]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hapon, M.B.; Goyeneche, A.A.; Srinivasan, R.; Gamarra-Luques, C.D.; Callegari, E.A.; Drappeau, D.D.; Terpstra, E.J.; Pan, B.; Knapp, J.R.; et al. Mifepristone increases mRNA translation rate, triggers the unfolded protein response, increases autophagic flux, and kills ovarian cancer cells in combination with proteasome or lysosome inhibitors. Mol. Oncol. 2016, 10, 1099–1117. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Motzer, R.J.; Tong, Y.; Bosl, G.J. Computerized quantitation of synergism and antagonism of taxol, topotecan, and cisplatin against human teratocarcinoma cell growth: A rational approach to clinical protocol design. J. Natl. Cancer Inst. 1994, 86, 1517–1524. [Google Scholar] [CrossRef]
- Gamarra-Luques, C.D.; Goyeneche, A.A.; Hapon, M.B.; Telleria, C.M. Mifepristone prevents repopulation of ovarian cancer cells escaping cisplatin-paclitaxel therapy. BMC Cancer 2012, 12, 200. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.R.; Noble, M.E.; Lawrie, A.M.; Morris, M.C.; Tunnah, P.; Divita, G.; Johnson, L.N.; Endicott, J.A. Effects of phosphorylation of threonine 160 on cyclin-dependent kinase 2 structure and activity. J. Biol. Chem. 1999, 274, 8746–8756. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Schonthal, A.H. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem. Pharmacol. 2013, 85, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Vauleon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef]
- Clarke, H.J.; Chambers, J.E.; Liniker, E.; Marciniak, S.J. Endoplasmic reticulum stress in malignancy. Cancer Cell 2014, 25, 563–573. [Google Scholar] [CrossRef]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect Biol. 2013, 5, a013169. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Bussink, J.; Sweep, F.C.; Span, P.N. The unfolded protein response as a target for cancer therapy. Biochim. Biophys. Acta 2014, 1846, 277–284. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Amaravadi, R.K. Targeting the unfolded protein response in cancer. Pharmacol. Res. 2017, 120, 258–266. [Google Scholar] [CrossRef]
- Miyake, H.; Hara, I.; Arakawa, S.; Kamidono, S. Stress protein GRP78 prevents apoptosis induced by calcium ionophore, ionomycin, but not by glycosylation inhibitor, tunicamycin, in human prostate cancer cells. J. Cell. Biochem. 2000, 77, 396–408. [Google Scholar] [CrossRef]
- Noda, I.; Fujieda, S.; Seki, M.; Tanaka, N.; Sunaga, H.; Ohtsubo, T.; Tsuzuki, H.; Fan, G.K.; Saito, H. Inhibition of N-linked glycosylation by tunicamycin enhances sensitivity to cisplatin in human head-and-neck carcinoma cells. Int. J. Cancer 1999, 80, 279–284. [Google Scholar] [CrossRef]
- Jensen, K.; Bikas, A.; Patel, A.; Kushchayeva, Y.; Costello, J.; McDaniel, D.; Burman, K.; Vasko, V. Nelfinavir inhibits proliferation and induces DNA damage in thyroid cancer cells. Endocr. Relat. Cancer 2017, 24, 147–156. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Al-Bari, M.A.A.; Ito, Y.; Ahmed, S.; Radwan, N.; Ahmed, H.S.; Eid, N. Targeting Autophagy with Natural Products as a Potential Therapeutic Approach for Cancer. Int. J. Mol. Sci. 2021, 22, 9806. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.C.; Sathiyaseelan, P.; Ho, C.; Gorski, S.M. Evolution of tools and methods for monitoring autophagic flux in mammalian cells. Biochem. Soc. Trans. 2018, 46, 97–110. [Google Scholar] [CrossRef]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Koromilas, A.E. Roles of the translation initiation factor eIF2alpha serine 51 phosphorylation in cancer formation and treatment. Biochim. Biophys. Acta 2015, 1849, 871–880. [Google Scholar] [CrossRef]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Jiang, Z.; Bernhard, E.J.; Evans, S.M.; Koch, C.J.; Hahn, S.M.; Maity, A. Nelfinavir down-regulates hypoxia-inducible factor 1alpha and VEGF expression and increases tumor oxygenation: Implications for radiotherapy. Cancer Res. 2006, 66, 9252–9259. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.E.; Milum, K.; Han, C.; Huang, Y.W.; Wani, G.; Thomale, J.; Wani, A.A. Differential contributory roles of nucleotide excision and homologous recombination repair for enhancing cisplatin sensitivity in human ovarian cancer cells. Mol. Cancer 2011, 10, 24. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van, I.W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Masoodi, T.; Siraj, S.; Siraj, A.K.; Azam, S.; Qadri, Z.; Parvathareddy, S.K.; Tulbah, A.; Al-Dayel, F.; AlHusaini, H.; AlOmar, O.; et al. Genetic heterogeneity and evolutionary history of high-grade ovarian carcinoma and matched distant metastases. Br. J. Cancer 2020, 122, 1219–1230. [Google Scholar] [CrossRef]
- Shih, I.M.; Wang, Y.; Wang, T.L. The Origin of Ovarian Cancer Species and Precancerous Landscape. Am. J. Pathol. 2021, 191, 26–39. [Google Scholar] [CrossRef]
- Bao, Y. Chromatin response to DNA double-strand break damage. Epigenomics 2011, 3, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Farnham, P.J. KAP1 protein: An enigmatic master regulator of the genome. J. Biol. Chem. 2011, 286, 26267. [Google Scholar] [CrossRef]
- Pyrko, P.; Kardosh, A.; Wang, W.; Xiong, W.; Schonthal, A.H.; Chen, T.C. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res. 2007, 67, 10920–10928. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Thomas, S.; Golden, E.B.; Gaffney, K.J.; Hofman, F.M.; Chen, T.C.; Louie, S.G.; Petasis, N.A.; Schonthal, A.H. Enhanced killing of chemo-resistant breast cancer cells via controlled aggravation of ER stress. Cancer Lett. 2009, 282, 87–97. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. That which does not kill me makes me stronger: Adapting to chronic ER stress. Trends Biochem. Sci. 2007, 32, 469–476. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Garg, A.D.; Agostinis, P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013, 332, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, S.; Gills, J.J.; Mercado-Matos, J.R.; Lopiccolo, J.; Wilson, W., III; Hollander, M.C.; Dennis, P.A. Synergistic effects of nelfinavir and bortezomib on proteotoxic death of NSCLC and multiple myeloma cells. Cell Death Dis. 2012, 3, e353. [Google Scholar] [CrossRef]
- Chakravarty, G.; Mathur, A.; Mallade, P.; Gerlach, S.; Willis, J.; Datta, A.; Srivastav, S.; Abdel-Mageed, A.B.; Mondal, D. Nelfinavir targets multiple drug resistance mechanisms to increase the efficacy of doxorubicin in MCF-7/Dox breast cancer cells. Biochimie 2016, 124, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Tamura, S.; Dubeau, L.; Mhawech-Fauceglia, P.; Miyagi, Y.; Kato, H.; Lieberman, R.; Buckanovich, R.J.; Lin, Y.G.; Neamati, N. Clinicopathological significance of endoplasmic reticulum stress proteins in ovarian carcinoma. Sci. Rep. 2020, 10, 2160. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Su, L.; Yuan, Y.C.; Li, H.; Chow, W.A. Nelfinavir and nelfinavir analogs block site-2 protease cleavage to inhibit castration-resistant prostate cancer. Sci. Rep. 2015, 5, 9698. [Google Scholar] [CrossRef]
- Gills, J.J.; Lopiccolo, J.; Dennis, P.A. Nelfinavir, a new.w anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy 2008, 4, 107–109. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Greshock, J.; Colligon, T.A.; Wang, Y.; Ward, R.; Katsaros, D.; Lassus, H.; Butzow, R.; Godwin, A.K.; et al. Frequent genetic abnormalities of the PI3K/AKT pathway in primary ovarian cancer predict patient outcome. Genes Chromosom. Cancer 2011, 50, 606–618. [Google Scholar] [CrossRef]
- Martins, F.C.; Santiago, I.; Trinh, A.; Xian, J.; Guo, A.; Sayal, K.; Jimenez-Linan, M.; Deen, S.; Driver, K.; Mack, M.; et al. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss as a common driver event and prognostic classifier. Genome Biol. 2014, 15, 526. [Google Scholar] [CrossRef]
- Dunn, G.P.; Cheung, H.W.; Agarwalla, P.K.; Thomas, S.; Zektser, Y.; Karst, A.M.; Boehm, J.S.; Weir, B.A.; Berlin, A.M.; Zou, L.; et al. In vivo multiplexed interrogation of amplified genes identifies GAB2 as an ovarian cancer oncogene. Proc. Natl. Acad. Sci. USA 2014, 111, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Soprano, M.; Sorriento, D.; Rusciano, M.R.; Maione, A.S.; Limite, G.; Forestieri, P.; D’Angelo, D.; D’Alessio, M.; Campiglia, P.; Formisano, P.; et al. Oxidative Stress Mediates the Antiproliferative Effects of Nelfinavir in Breast Cancer Cells. PLoS ONE 2016, 11, e0155970. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Johnson, C.E.; Wiltshire, M.; Errington, R.J.; Tee, A.R. Targeting protein homeostasis with nelfinavir/salinomycin dual therapy effectively induces death of mTORC1 hyperactive cells. Oncotarget 2017, 8, 48711–48724. [Google Scholar] [CrossRef]
- Kraus, M.; Bader, J.; Overkleeft, H.; Driessen, C. Nelfinavir augments proteasome inhibition by bortezomib in myeloma cells and overcomes bortezomib and carfilzomib resistance. Blood Cancer J. 2013, 3, e103. [Google Scholar] [CrossRef]
- Kraus, M.; Muller-Ide, H.; Ruckrich, T.; Bader, J.; Overkleeft, H.; Driessen, C. Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leuk. Res. 2014, 38, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Meier-Stephenson, V.; Riemer, J.; Narendran, A. The HIV protease inhibitor, nelfinavir, as a novel therapeutic approach for the treatment of refractory pediatric leukemia. Onco. Targets Ther. 2017, 10, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Petrich, A.M.; Leshchenko, V.; Kuo, P.Y.; Xia, B.; Thirukonda, V.K.; Ulahannan, N.; Gordon, S.; Fazzari, M.J.; Ye, B.H.; Sparano, J.A.; et al. Akt inhibitors MK-2206 and nelfinavir overcome mTOR inhibitor resistance in diffuse large B-cell lymphoma. Clin. Cancer Res. 2012, 18, 2534–2544. [Google Scholar] [CrossRef]
- Yang, Y.; Ikezoe, T.; Takeuchi, T.; Adachi, Y.; Ohtsuki, Y.; Takeuchi, S.; Koeffler, H.P.; Taguchi, H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human prostate cancer LNCaP cells in vitro and in vivo in conjunction with blockade of androgen receptor STAT3 and AKT signaling. Cancer Sci. 2005, 96, 425–433. [Google Scholar] [CrossRef]
- Yang, Y.; Ikezoe, T.; Nishioka, C.; Bandobashi, K.; Takeuchi, T.; Adachi, Y.; Kobayashi, M.; Takeuchi, S.; Koeffler, H.P.; Taguchi, H. NFV, an HIV-1 protease inhibitor, induces growth arrest, reduced Akt signalling, apoptosis and docetaxel sensitisation in NSCLC cell lines. Br. J. Cancer 2006, 95, 1653–1662. [Google Scholar] [CrossRef]
- Gupta, A.K.; Cerniglia, G.J.; Mick, R.; McKenna, W.G.; Muschel, R.J. HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res. 2005, 65, 8256–8265. [Google Scholar] [CrossRef]
- Jiang, Z.; Pore, N.; Cerniglia, G.J.; Mick, R.; Georgescu, M.M.; Bernhard, E.J.; Hahn, S.M.; Gupta, A.K.; Maity, A. Phosphatase and tensin homologue deficiency in glioblastoma confers resistance to radiation and temozolomide that is reversed by the protease inhibitor nelfinavir. Cancer Res. 2007, 67, 4467–4473. [Google Scholar] [CrossRef]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Maity, A. HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia 2006, 8, 889–895. [Google Scholar] [CrossRef]
- Plastaras, J.P.; Vapiwala, N.; Ahmed, M.S.; Gudonis, D.; Cerniglia, G.J.; Feldman, M.D.; Frank, I.; Gupta, A.K. Validation and toxicity of PI3K/Akt pathway inhibition by HIV protease inhibitors in humans. Cancer Biol. Ther. 2008, 7, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Meng, Q.; Sun, Y.; Wang, C.; Huo, X.; Liu, Z.; Sun, P.; Sun, H.; Ma, X.; Liu, K. Targeting P-Glycoprotein: Nelfinavir Reverses Adriamycin Resistance in K562/ADR Cells. Cell. Physiol. Biochem. 2018, 51, 1616–1631. [Google Scholar] [CrossRef]
- Ikezoe, T.; Saito, T.; Bandobashi, K.; Yang, Y.; Koeffler, H.P.; Taguchi, H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of signal transducer and activator of transcription 3 and extracellular signal-regulated kinase 1/2. Mol. Cancer Ther. 2004, 3, 473–479. [Google Scholar]
- Kushchayeva, Y.; Jensen, K.; Recupero, A.; Costello, J.; Patel, A.; Klubo-Gwiezdzinska, J.; Boyle, L.; Burman, K.; Vasko, V. The HIV protease inhibitor nelfinavir down-regulates RET signaling and induces apoptosis in medullary thyroid cancer cells. J. Clin. Endocrinol. Metab. 2014, 99, E734–E745. [Google Scholar] [CrossRef]
- Gupta, A.K.; Wilke, W.W.; Taylor, E.N.; Bodeker, K.L.; Hoffman, H.T.; Milhem, M.M.; Buatti, J.M.; Robinson, R.A. Signaling pathways in adenoid cystic cancers: Implications for treatment. Cancer Biol. Ther. 2009, 8, 1947–1951. [Google Scholar] [CrossRef]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Jacquemont, C.; Chandramohan, K.V.; Couch, F.J.; Langdon, S.P.; Wurz, K.; Higgins, J.; Villegas, E.; Taniguchi, T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009, 69, 6381–6386. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Caruso, D.; Strudel, M.; Tomao, S.; Tomao, F. Update on Poly-ADP-ribose polymerase inhibition for ovarian cancer treatment. J. Transl. Med. 2016, 14, 267. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Bruning, A.; Vogel, M.; Mylonas, I.; Friese, K.; Burges, A. Bortezomib targets the caspase-like proteasome activity in cervical cancer cells, triggering apoptosis that can be enhanced by nelfinavir. Curr. Cancer Drug Targets 2011, 11, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Chen, R.; Chen, J.; Qi, Q.; Pan, Y.; Du, L.; Xiao, G.; Jiang, S. Combining metformin and nelfinavir exhibits synergistic effects against the growth of human cervical cancer cells and xenograft in nude mice. Sci. Rep. 2017, 7, 43373. [Google Scholar] [CrossRef] [PubMed]
- Kattel, K.; Evande, R.; Tan, C.; Mondal, G.; Grem, J.L.; Mahato, R.I. Impact of CYP2C19 polymorphism on the pharmacokinetics of nelfinavir in patients with pancreatic cancer. Br. J. Clin. Pharmacol. 2015, 80, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic-Ivanic, D.; Fagone, P.; McCubrey, J.; Bendtzen, K.; Mijatovic, S.; Nicoletti, F. HIV-protease inhibitors for the treatment of cancer: Repositioning HIV protease inhibitors while developing more potent NO-hybridized derivatives? Int. J. Cancer 2017, 140, 1713–1726. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 (µM) |
|---|---|
| PEO1 | 0.56 ± 0.08 |
| PEO14 | 0.65 ± 0.06 |
| PEO23 | 3.36 ± 0.14 |
| PEO4 | 6.79 ± 0.51 |
| PEO6 | 8.66 ± 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subeha, M.R.; Goyeneche, A.A.; Bustamante, P.; Lisio, M.A.; Burnier, J.V.; Telleria, C.M. Nelfinavir Induces Cytotoxicity towards High-Grade Serous Ovarian Cancer Cells, Involving Induction of the Unfolded Protein Response, Modulation of Protein Synthesis, DNA Damage, Lysosomal Impairment, and Potentiation of Toxicity Caused by Proteasome Inhibition. Cancers 2022, 14, 99. https://doi.org/10.3390/cancers14010099
Subeha MR, Goyeneche AA, Bustamante P, Lisio MA, Burnier JV, Telleria CM. Nelfinavir Induces Cytotoxicity towards High-Grade Serous Ovarian Cancer Cells, Involving Induction of the Unfolded Protein Response, Modulation of Protein Synthesis, DNA Damage, Lysosomal Impairment, and Potentiation of Toxicity Caused by Proteasome Inhibition. Cancers. 2022; 14(1):99. https://doi.org/10.3390/cancers14010099
Chicago/Turabian StyleSubeha, Mahbuba R., Alicia A. Goyeneche, Prisca Bustamante, Michael A. Lisio, Julia V. Burnier, and Carlos M. Telleria. 2022. "Nelfinavir Induces Cytotoxicity towards High-Grade Serous Ovarian Cancer Cells, Involving Induction of the Unfolded Protein Response, Modulation of Protein Synthesis, DNA Damage, Lysosomal Impairment, and Potentiation of Toxicity Caused by Proteasome Inhibition" Cancers 14, no. 1: 99. https://doi.org/10.3390/cancers14010099
APA StyleSubeha, M. R., Goyeneche, A. A., Bustamante, P., Lisio, M. A., Burnier, J. V., & Telleria, C. M. (2022). Nelfinavir Induces Cytotoxicity towards High-Grade Serous Ovarian Cancer Cells, Involving Induction of the Unfolded Protein Response, Modulation of Protein Synthesis, DNA Damage, Lysosomal Impairment, and Potentiation of Toxicity Caused by Proteasome Inhibition. Cancers, 14(1), 99. https://doi.org/10.3390/cancers14010099