Current and Emerging Approaches to Study Microenvironmental Interactions and Drug Activity in Classical Hodgkin Lymphoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
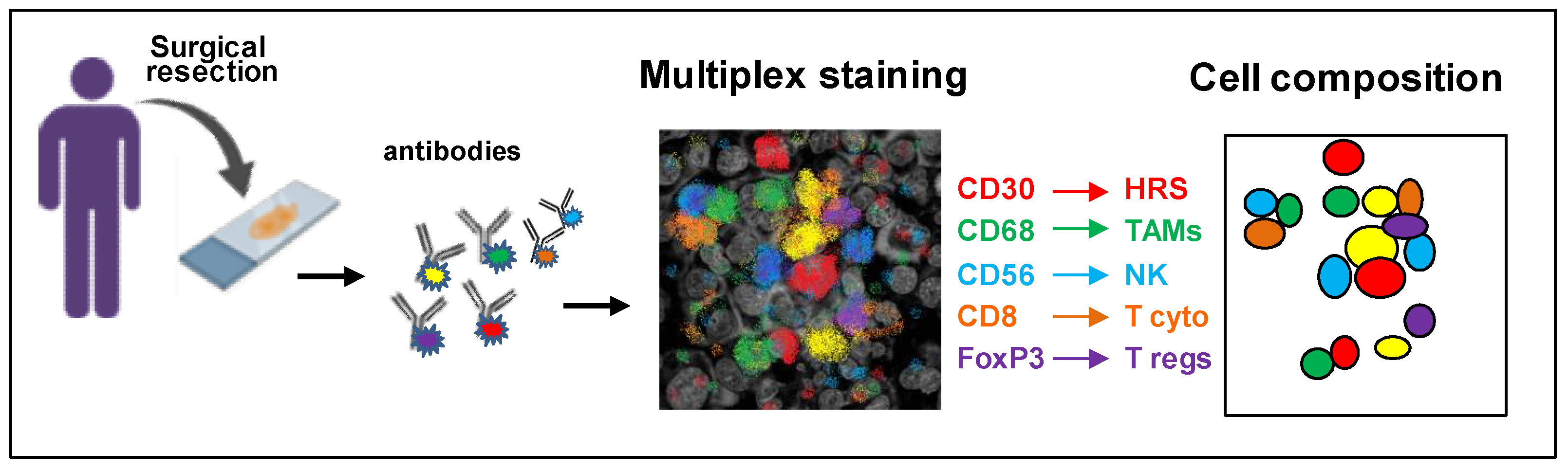
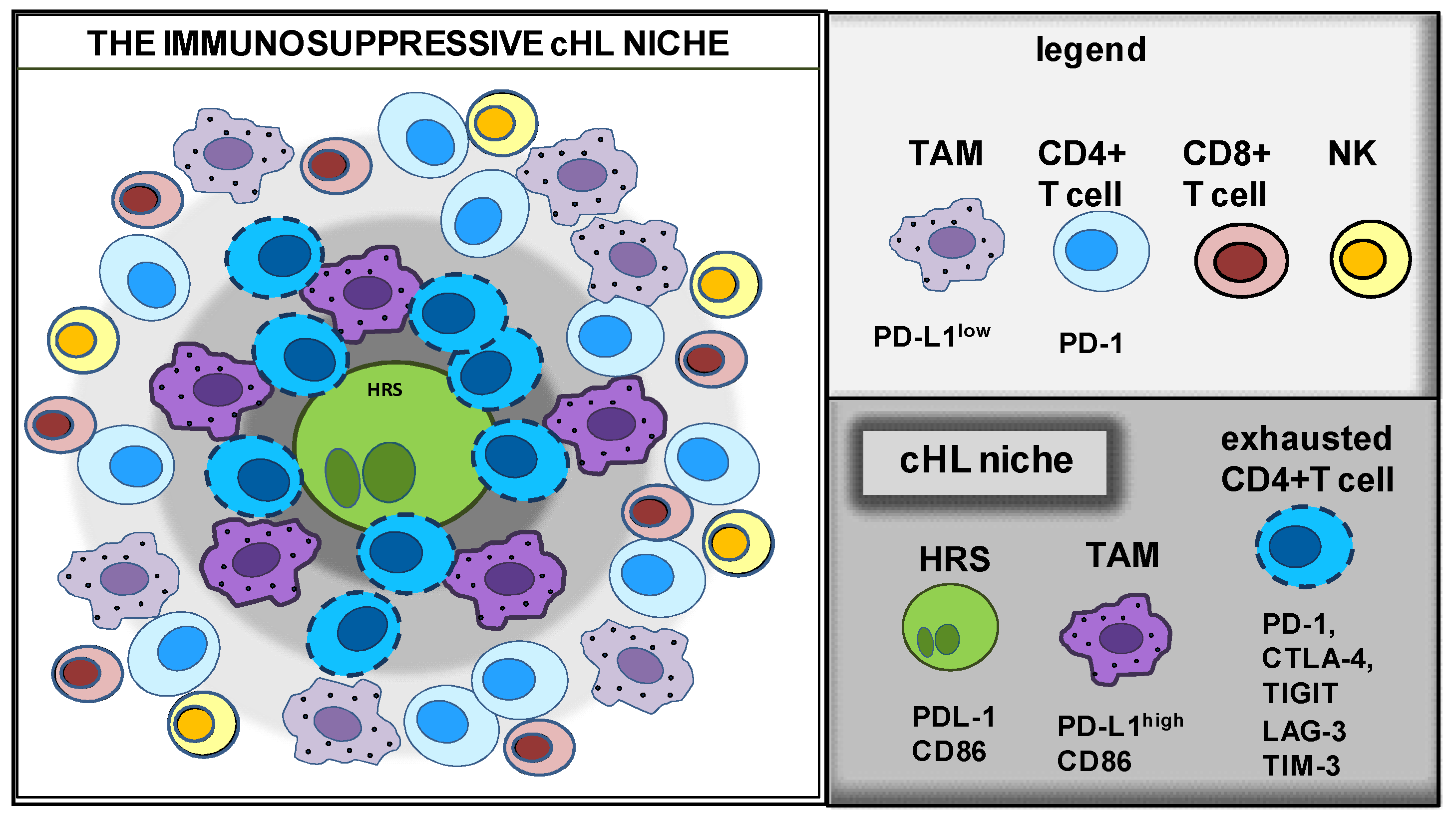
2. Characterization of the TME Composition
Identifying the Immunoprotective cHL Niche with Multiplex Platforms
3. Identify the State of the Disease with Gene Expression Profiling of the TME
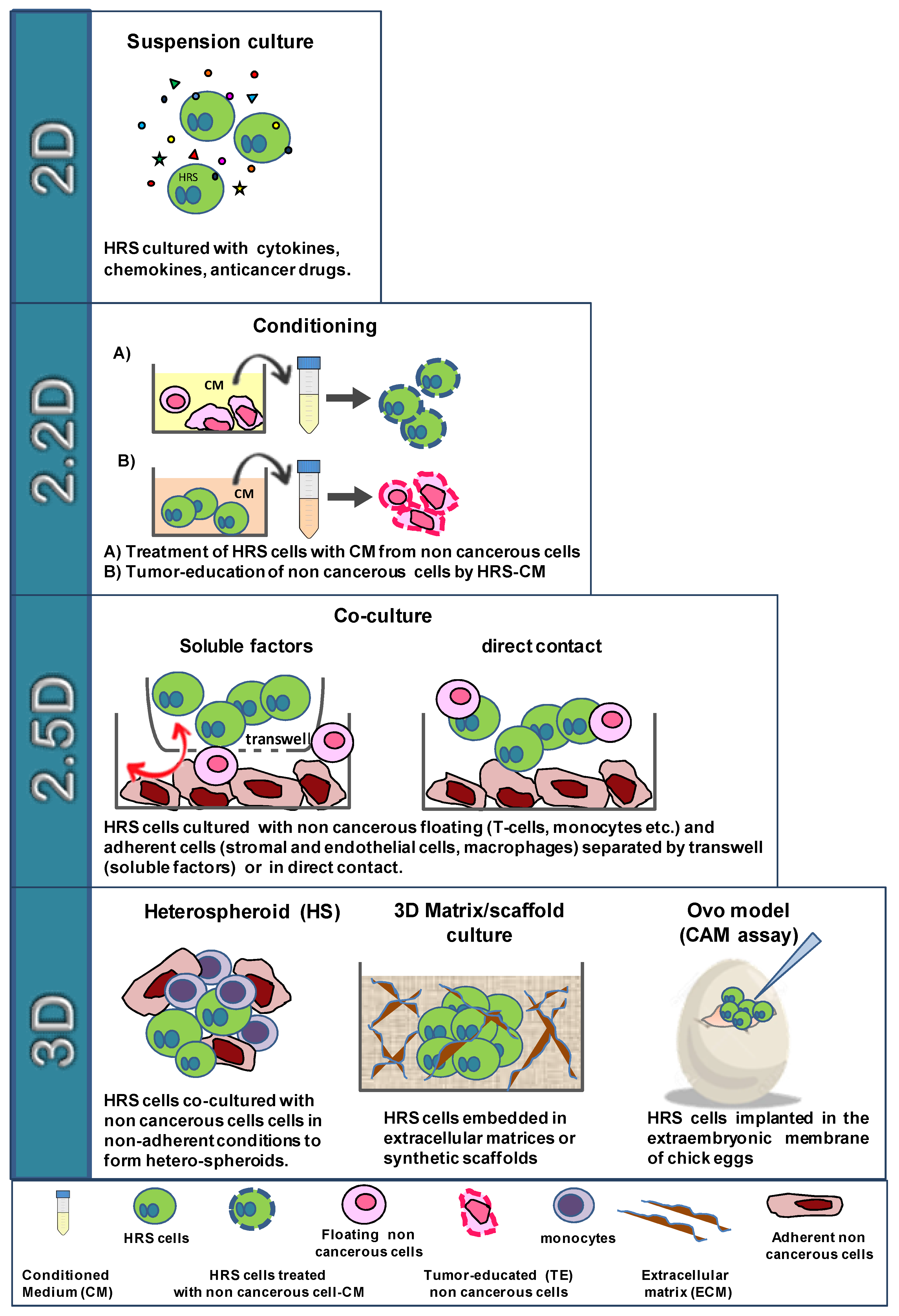
4. In Vitro Models to Study HRS/TME Interactions
4.1. The Cross-Talk of HRS Cells with T lymphocytes
4.2. The Cross-Talk of HRS Cells with Eosinophils
4.3. The Cross-Talk of HRS Cells with Monocytes
4.4. The Cross-Talk of HRS Cells with Fibroblasts and MSCs
5. Extracellular Vesicles in HRS-TME Communication and Immune Suppression
6. Trogocytosis in the Formation of the Immunosuppressive TME
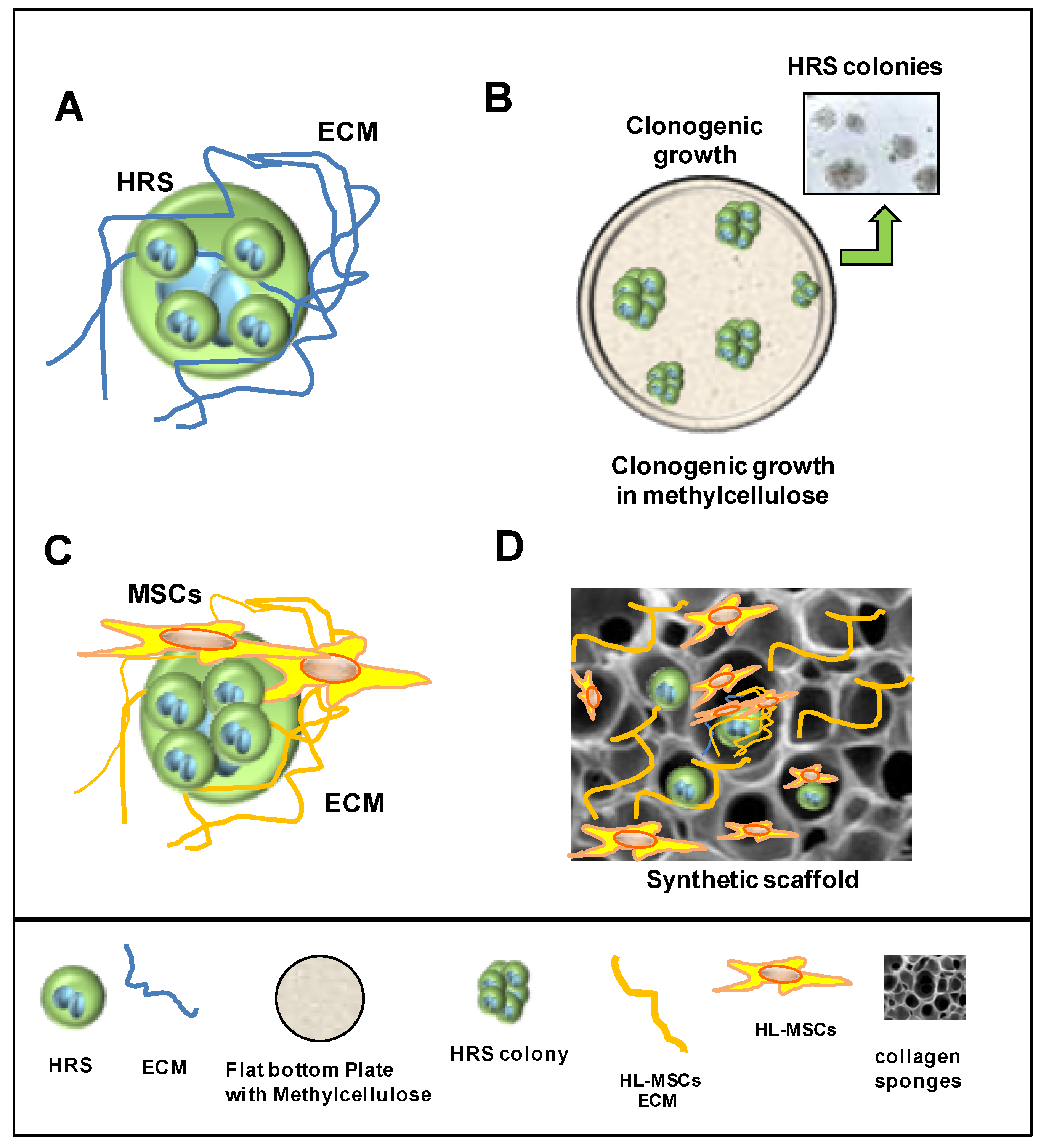
7. 3D Models to Study Tumor–Stroma Interactions and Drug Activity
7.1. Cultivation in 3D Extracellular Matrix
7.2. Heterospheroids (HS) to Study the Interactions of HRS Cells with TME and Drug Activity
7.2.1. Heterospheroids (HS) to Study ADAM10 Inhibitors in Combination with BV
7.2.2. Heterospheroids (HS) to Study the TME-Protective Effects against Drugs
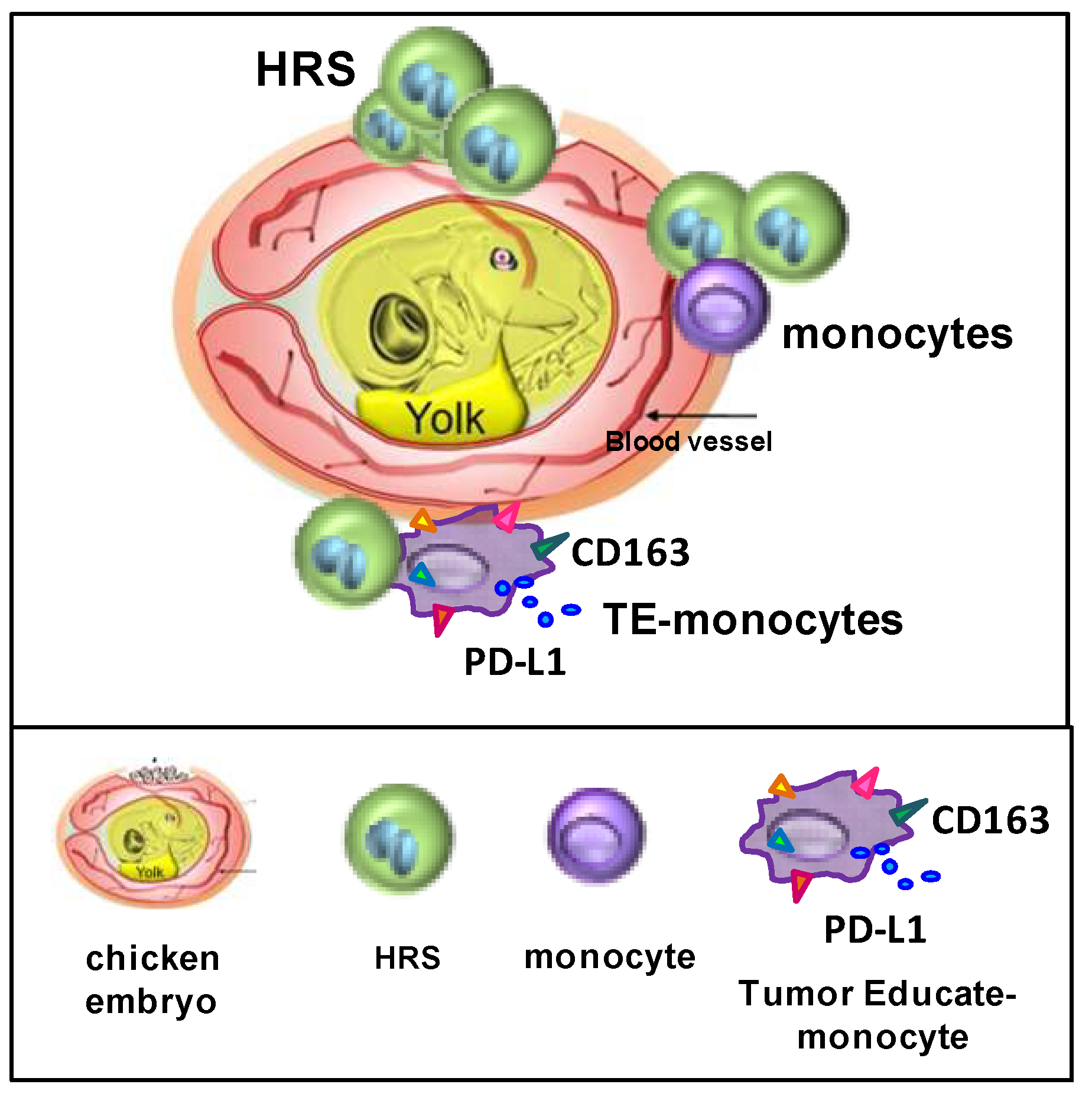
8. The CAM Model to Study Dissemination and Interactions of HRSs with Monocytes
9. In Vivo Tumor Xenograft to Study TME Interactions
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Connors, J.M.; Cozen, W.; Steidl, C.; Carbone, A.; Hoppe, R.T.; Flechtner, H.-H.; Bartlett, N.L. Hodgkin Lymphoma. Nat. Rev. Dis. Primers 2020, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M. Hodgkin Lymphoma: A 2020 Update on Diagnosis, Risk-Stratification, and Management. Am. J. Hematol. 2020, 95, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.A.; Küppers, R. Molecular Biology of Hodgkin Lymphoma. Leukemia 2021, 35, 968–981. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Celegato, M.; Casagrande, N. Microenvironmental Interactions in Classical Hodgkin Lymphoma and Their Role in Promoting Tumor Growth, Immune Escape and Drug Resistance. Cancer Lett. 2016, 380, 243–252. [Google Scholar] [CrossRef]
- Veldman, J.; Visser, L.; Huberts-Kregel, M.; Muller, N.; Hepkema, B.; van den Berg, A.; Diepstra, A. Rosetting T Cells in Hodgkin Lymphoma Are Activated by Immunological Synapse Components HLA Class II and CD58. Blood 2020, 136, 2437–2441. [Google Scholar] [CrossRef]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T.; et al. Single-Cell Transcriptome Analysis Reveals Disease-Defining T-Cell Subsets in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.S.; Weirather, J.L.; Lipschitz, M.; Lako, A.; Chen, P.-H.; Griffin, G.K.; Armand, P.; Shipp, M.A.; Rodig, S.J. The Microenvironmental Niche in Classic Hodgkin Lymphoma Is Enriched for CTLA-4-Positive T Cells That Are PD-1-Negative. Blood 2019, 134, 2059–2069. [Google Scholar] [CrossRef]
- Englund, A.; Molin, D.; Enblad, G.; Karlén, J.; Glimelius, I.; Ljungman, G.; Amini, R.-M. The Role of Tumour-Infiltrating Eosinophils, Mast Cells and Macrophages in Classical and Nodular Lymphocyte Predominant Hodgkin Lymphoma in Children. Eur. J. Haematol. 2016, 97, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-Associated Macrophages and Survival in Classic Hodgkin’s Lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Vardhana, S.; Younes, A. The Immune Microenvironment in Hodgkin Lymphoma: T Cells, B Cells, and Immune Checkpoints. Haematologica 2016, 101, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Andersen, M.D.; Kamper, P.; Nielsen, P.S.; Bendix, K.; Riber-Hansen, R.; Steiniche, T.; Hamilton-Dutoit, S.; Clausen, M.; d’Amore, F. Tumour-Associated Mast Cells in Classical Hodgkin’s Lymphoma: Correlation with Histological Subtype, Other Tumour-Infiltrating Inflammatory Cell Subsets and Outcome. Eur. J. Haematol. 2016, 96, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Yokote, T.; Hiraoka, N.; Nishiwaki, U.; Hanafusa, T.; Nishimura, Y.; Tsuji, M. Role of Mast Cells in Fibrosis of Classical Hodgkin Lymphoma. Int. J. Immunopathol. Pharmacol. 2016, 29, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Gholiha, A.R.; Hollander, P.; Hedstrom, G.; Sundstrom, C.; Molin, D.; Smedby, K.E.; Hjalgrim, H.; Glimelius, I.; Amini, R.-M.; Enblad, G. High Tumour Plasma Cell Infiltration Reflects an Important Microenvironmental Component in Classic Hodgkin Lymphoma Linked to Presence of B-Symptoms. Br. J. Haematol. 2019, 184, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, H.P.; Paes Leme, A.F.; Hallek, M. Role of ADAM10 as a CD30 Sheddase in Classical Hodgkin Lymphoma. Front. Immunol. 2020, 11, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankov, K.; Döring, C.; Ustaszewski, A.; Giefing, M.; Herling, M.; Cencioni, C.; Spallotta, F.; Gaetano, C.; Küppers, R.; Hansmann, M.-L.; et al. Fibroblasts in Nodular Sclerosing Classical Hodgkin Lymphoma Are Defined by a Specific Phenotype and Protect Tumor Cells from Brentuximab-Vedotin Induced Injury. Cancers 2019, 11, 1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poggi, A.; Musso, A.; Dapino, I.; Zocchi, M.R. Mechanisms of Tumor Escape from Immune System: Role of Mesenchymal Stromal Cells. Immunol. Lett. 2014, 159, 55–72. [Google Scholar] [CrossRef]
- Linke, F.; Harenberg, M.; Nietert, M.M.; Zaunig, S.; von Bonin, F.; Arlt, A.; Szczepanowski, M.; Weich, H.A.; Lutz, S.; Dullin, C.; et al. Microenvironmental Interactions between Endothelial and Lymphoma Cells: A Role for the Canonical WNT Pathway in Hodgkin Lymphoma. Leukemia 2017, 31, 361–372. [Google Scholar] [CrossRef]
- Arlt, A.; von Bonin, F.; Rehberg, T.; Perez-Rubio, P.; Engelmann, J.C.; Limm, K.; Reinke, S.; Dullin, C.; Sun, X.; Specht, R.; et al. High CD206 Levels in Hodgkin Lymphoma-Educated Macrophages Are Linked to Matrix-Remodeling and Lymphoma Dissemination. Mol. Oncol. 2020, 14, 571–589. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Borghese, C.; Casagrande, N. Formation of the Immunosuppressive Microenvironment of Classic Hodgkin Lymphoma and Therapeutic Approaches to Counter It. Int. J. Mol. Sci. 2019, 20, 2416. [Google Scholar] [CrossRef] [Green Version]
- Carey, C.D.; Gusenleitner, D.; Lipschitz, M.; Roemer, M.G.M.; Stack, E.C.; Gjini, E.; Hu, X.; Redd, R.; Freeman, G.J.; Neuberg, D.; et al. Topological Analysis Reveals a PD-L1-Associated Microenvironmental Niche for Reed-Sternberg Cells in Hodgkin Lymphoma. Blood 2017, 130, 2420–2430. [Google Scholar] [CrossRef]
- Mottok, A.; Steidl, C. Biology of Classical Hodgkin Lymphoma: Implications for Prognosis and Novel Therapies. Blood 2018, 131, 1654–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, A.; Gloghini, A.; Gattei, V.; Aldinucci, D.; Degan, M.; De Paoli, P.; Zagonel, V.; Pinto, A. Expression of Functional CD40 Antigen on Reed-Sternberg Cells and Hodgkin’s Disease Cell Lines. Blood 1995, 85, 780–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wein, F.; Weniger, M.A.; Höing, B.; Arnolds, J.; Hüttmann, A.; Hansmann, M.-L.; Hartmann, S.; Küppers, R. Complex Immune Evasion Strategies in Classical Hodgkin Lymphoma. Cancer Immunol. Res. 2017, 5, 1122–1132. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.G.; Young, L.S. An Etiological Role for the Epstein-Barr Virus in the Pathogenesis of Classical Hodgkin Lymphoma. Blood 2019, 134, 591–596. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Caruso, A.; De Paoli, P.; Dolcetti, R. The Impact of EBV and HIV Infection on the Microenvironmental Niche Underlying Hodgkin Lymphoma Pathogenesis. Int. J. Cancer 2017, 140, 1233–1245. [Google Scholar] [CrossRef] [PubMed]
- Cader, F.Z.; Vockerodt, M.; Bose, S.; Nagy, E.; Brundler, M.-A.; Kearns, P.; Murray, P.G. The EBV Oncogene LMP1 Protects Lymphoma Cells from Cell Death through the Collagen-Mediated Activation of DDR1. Blood 2013, 122, 4237–4245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumforth, K.R.N.; Birgersdotter, A.; Reynolds, G.M.; Wei, W.; Kapatai, G.; Flavell, J.R.; Kalk, E.; Piper, K.; Lee, S.; Machado, L.; et al. Expression of the Epstein-Barr Virus-Encoded Epstein-Barr Virus Nuclear Antigen 1 in Hodgkin’s Lymphoma Cells Mediates Up-Regulation of CCL20 and the Migration of Regulatory T Cells. Am. J. Pathol. 2008, 173, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrzalikova, K.; Pugh, M.; Mundo, L.; Murray, P. The Contribution of Ebv to the Pathogenesis of Classical Hodgkin Lymphoma. Ann. Lymphoma 2021, 5, 30. [Google Scholar] [CrossRef]
- Navarro, J.-T.; Moltó, J.; Tapia, G.; Ribera, J.-M. Hodgkin Lymphoma in People Living with HIV. Cancers 2021, 13, 4366. [Google Scholar] [CrossRef]
- Hartmann, S.; Jakobus, C.; Rengstl, B.; Döring, C.; Newrzela, S.; Brodt, H.-R.; Wolf, T.; Hansmann, M.-L. Spindle-Shaped CD163+ Rosetting Macrophages Replace CD4+ T-Cells in HIV-Related Classical Hodgkin Lymphoma. Mod. Pathol. 2013, 26, 648–657. [Google Scholar] [CrossRef]
- Advani, R.H.; Moskowitz, A.J.; Bartlett, N.L.; Vose, J.M.; Ramchandren, R.; Feldman, T.A.; LaCasce, A.S.; Christian, B.A.; Ansell, S.M.; Moskowitz, C.H.; et al. Brentuximab Vedotin in Combination with Nivolumab in Relapsed or Refractory Hodgkin Lymphoma: 3-Year Study Results. Blood 2021, 138, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.F.; Burton, C.; Radford, J.; Miall, F.; Townsend, W.; Santoro, A.; Zinzani, P.L.; Lewis, D.; Fowst, C.; Brar, S.; et al. Avelumab in Relapsed/Refractory Classical Hodgkin Lymphoma: Phase 1b Results from the JAVELIN Hodgkins Trial. Blood Adv. 2021, 5, 3387–3396. [Google Scholar] [CrossRef]
- Ho, C.; Ruella, M.; Levine, B.L.; Svoboda, J. Adoptive T-Cell Therapy for Hodgkin Lymphoma. Blood Adv. 2021, 5, 4291–4302. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Grover, N.S.; Beaven, A.W.; Lulla, P.D.; Wu, M.-F.; Ivanova, A.; Wang, T.; Shea, T.C.; Rooney, C.M.; Dittus, C.; et al. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. J. Clin. Oncol. 2020, 38, 3794–3804. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; He, S.; Zhu, Y.; Yu, W.; Yang, D.; Zhao, X. Humanized CD30-Targeted Chimeric Antigen Receptor T Cells Exhibit Potent Preclinical Activity Against Hodgkin’s Lymphoma Cells. Front. Cell Dev. Biol. 2021, 9, 775599. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, S.L.; Careddu, G.; Serio, S.; Consonni, F.M.; Maeda, A.; Viswanadha, S.; Vakkalanka, S.; Castagna, L.; Santoro, A.; Allavena, P.; et al. Targeting Cancer Cells and Tumor Microenvironment in Preclinical and Clinical Models of Hodgkin Lymphoma Using the Dual PI3Kδ/γ Inhibitor RP6530. Clin. Cancer Res. 2019, 25, 1098–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casagrande, N.; Borghese, C.; Favero, A.; Vicenzetto, C.; Aldinucci, D. Trabectedin Overcomes Doxorubicin-Resistance, Counteracts Tumor-Immunosuppressive Reprogramming of Monocytes and Decreases Xenograft Growth in Hodgkin Lymphoma. Cancer Lett. 2021, 500, 182–193. [Google Scholar] [CrossRef]
- Chen, R.; Herrera, A.F.; Hou, J.; Chen, L.; Wu, J.; Guo, Y.; Synold, T.W.; Ngo, V.N.; Puverel, S.; Mei, M.; et al. Inhibition of MDR1 Overcomes Resistance to Brentuximab Vedotin in Hodgkin Lymphoma. Clin. Cancer Res. 2020, 26, 1034–1044. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Loganzo, F.; Sung, M.; Gerber, H.-P. Mechanisms of Resistance to Antibody-Drug Conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Lin, Y.; Song, Z.; Xiao, W.; Chen, L.; Yin, J.; Zhou, Y.; Barta, S.K.; Petrus, M.; Waldmann, T.A.; et al. A20 and RBX1 Regulate Brentuximab Vedotin Sensitivity in Hodgkin Lymphoma Models. Clin. Cancer Res. 2020, 26, 4093–4106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honoré, B.; Andersen, M.D.; Wilken, D.; Kamper, P.; d’Amore, F.; Hamilton-Dutoit, S.; Ludvigsen, M. Classic Hodgkin Lymphoma Refractory for ABVD Treatment Is Characterized by Pathologically Activated Signal Transduction Pathways as Revealed by Proteomic Profiling. Cancers 2022, 14, 247. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.M.; Asselin-Labat, M.-L.; Nguyen, Q.; Berthelet, J.; Tan, X.; Wimmer, V.C.; Merino, D.; Rogers, K.L.; Naik, S.H. Spatial Omics and Multiplexed Imaging to Explore Cancer Biology. Nat. Methods 2021, 18, 997–1012. [Google Scholar] [CrossRef] [PubMed]
- Remark, R.; Merghoub, T.; Grabe, N.; Litjens, G.; Damotte, D.; Wolchok, J.D.; Merad, M.; Gnjatic, S. In-Depth Tissue Profiling Using Multiplexed Immunohistochemical Consecutive Staining on Single Slide. Sci. Immunol. 2016, 1, aaf6925. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; Wang, C.; Roman, K.A.; Hoyt, C.C. Multiplexed Immunohistochemistry, Imaging, and Quantitation: A Review, with an Assessment of Tyramide Signal Amplification, Multispectral Imaging and Multiplex Analysis. Methods 2014, 70, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Wharton, K.A.; Wood, D.; Manesse, M.; Maclean, K.H.; Leiss, F.; Zuraw, A. Tissue Multiplex Analyte Detection in Anatomic Pathology - Pathways to Clinical Implementation. Front. Mol. Biosci. 2021, 8, 672531. [Google Scholar] [CrossRef] [PubMed]
- Parra, E.R. Methods to Determine and Analyze the Cellular Spatial Distribution Extracted From Multiplex Immunofluorescence Data to Understand the Tumor Microenvironment. Front. Mol. Biosci. 2021, 8, 668340. [Google Scholar] [CrossRef]
- Lin, J.-R.; Izar, B.; Wang, S.; Yapp, C.; Mei, S.; Shah, P.M.; Santagata, S.; Sorger, P.K. Highly Multiplexed Immunofluorescence Imaging of Human Tissues and Tumors Using T-CyCIF and Conventional Optical Microscopes. Elife 2018, 7, e31657. [Google Scholar] [CrossRef]
- Hollman-Hewgley, D.; Lazare, M.; Bordwell, A.; Zebadua, E.; Tripathi, P.; Ross, A.S.; Fisher, D.; Adams, A.; Bouman, D.; O’Malley, D.P.; et al. A Single Slide Multiplex Assay for the Evaluation of Classical Hodgkin Lymphoma. Am. J. Surg. Pathol. 2014, 38, 1193–1202. [Google Scholar] [CrossRef]
- Rovira-Clavé, X.; Jiang, S.; Bai, Y.; Zhu, B.; Barlow, G.; Bhate, S.; Coskun, A.F.; Han, G.; Ho, C.-M.K.; Hitzman, C.; et al. Subcellular Localization of Biomolecules and Drug Distribution by High-Definition Ion Beam Imaging. Nat. Commun. 2021, 12, 4628. [Google Scholar] [CrossRef]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass Cytometry: Technique for Real Time Single Cell Multitarget Immunoassay Based on Inductively Coupled Plasma Time-of-Flight Mass Spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Goltsev, Y.; Samusik, N.; Kennedy-Darling, J.; Bhate, S.; Hale, M.; Vazquez, G.; Black, S.; Nolan, G.P. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 2018, 174, 968–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex Digital Spatial Profiling of Proteins and RNA in Fixed Tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Beechem, J.M. High-Plex Spatially Resolved RNA and Protein Detection Using Digital Spatial Profiling: A Technology Designed for Immuno-Oncology Biomarker Discovery and Translational Research. Methods Mol. Biol. 2020, 2055, 563–583. [Google Scholar] [CrossRef] [PubMed]
- Cornish, T.C. Clinical Application of Image Analysis in Pathology. Adv. Anat. Pathol. 2020, 27, 227–235. [Google Scholar] [CrossRef]
- Pugh, M.; Akarka, A.U.; Hunter, K.; Dojcinov, S.; Marafioti, T. Multiplex Immunohistochemistry in Lymphoma Pathology: A Research Tool for Study of the Immune Microenvironment. Diagn. Histopathol. 2020, 26, 407–420. [Google Scholar] [CrossRef]
- Roemer, M.G.M.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [Green Version]
- Roemer, M.G.M.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin Lymphoma with Reduced Β2M/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol. Res. 2016, 4, 910–916. [Google Scholar] [CrossRef] [Green Version]
- Au, Q.; Fang, J.; Juncker-Jensen, A.; Kuo, J.; Leones, E.; Sahafi, F.; Padmanabhan, R.; Hoe, N.; William, J. Characterization of Myeloid-Derived Suppressor Cells and Tumor Associated Macrophages Using MultiOmyxTM Hyperplexed Immunofluorescence Assay in Hodgkin Lymphoma. Blood 2018, 132, 4135. [Google Scholar] [CrossRef]
- Bolm, L.; Petruch, N.; Sivakumar, S.; Annels, N.E.; Frampton, A.E. Gene of the Month: T-Cell Immunoreceptor with Immunoglobulin and ITIM Domains (TIGIT). J. Clin. Pathol. 2022, 75, 217–221. [Google Scholar] [CrossRef]
- Li, W.; Blessin, N.C.; Simon, R.; Kluth, M.; Fischer, K.; Hube-Magg, C.; Makrypidi-Fraune, G.; Wellge, B.; Mandelkow, T.; Debatin, N.F.; et al. Expression of the Immune Checkpoint Receptor TIGIT in Hodgkin’s Lymphoma. BMC Cancer 2018, 18, 1209. [Google Scholar] [CrossRef] [PubMed]
- Annibali, O.; Bianchi, A.; Grifoni, A.; Tomarchio, V.; Tafuri, M.; Verri, M.; Avvisati, G.; Crescenzi, A. A Novel Scoring System for TIGIT Expression in Classic Hodgkin Lymphoma. Sci. Rep. 2021, 11, 7059. [Google Scholar] [CrossRef] [PubMed]
- El Halabi, L.; Adam, J.; Gravelle, P.; Marty, V.; Danu, A.; Lazarovici, J.; Ribrag, V.; Bosq, J.; Camara-Clayette, V.; Laurent, C.; et al. Expression of the Immune Checkpoint Regulators LAG-3 and TIM-3 in Classical Hodgkin Lymphoma. Clin. Lymphoma Myeloma Leuk. 2021, 21, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Karihtala, K.; Leivonen, S.-K.; Brück, O.; Karjalainen-Lindsberg, M.-L.; Mustjoki, S.; Pellinen, T.; Leppä, S. Prognostic Impact of Tumor-Associated Macrophages on Survival Is Checkpoint Dependent in Classical Hodgkin Lymphoma. Cancers 2020, 12, 877. [Google Scholar] [CrossRef] [Green Version]
- Roussel, M.; Lhomme, F.; Roe, C.E.; Bartkowiak, T.; Gravelle, P.; Laurent, C.; Fest, T.; Irish, J.M. Mass Cytometry Defines Distinct Immune Profile in Germinal Center B-Cell Lymphomas. Cancer Immunol. Immunother. 2020, 69, 407–420. [Google Scholar] [CrossRef]
- Jachimowicz, R.D.; Pieper, L.; Reinke, S.; Gontarewicz, A.; Plütschow, A.; Haverkamp, H.; Frauenfeld, L.; Fend, F.; Overkamp, M.; Jochims, F.; et al. Whole-Slide Image Analysis of the Tumor Microenvironment Identifies Low B-Cell Content as a Predictor of Adverse Outcome in Patients with Advanced-Stage Classical Hodgkin Lymphoma Treated with BEACOPP. Haematologica 2021, 106, 1684–1692. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next Generation of Immune Checkpoint Inhibitors and Beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- López-Pereira, B.; Fernández-Velasco, A.A.; Fernández-Vega, I.; Corte-Torres, D.; Quirós, C.; Villegas, J.A.; Palomo, P.; González, S.; González, A.P.; Payer, Á.; et al. Expression of CD47 Antigen in Reed-Sternberg Cells as a New Potential Biomarker for Classical Hodgkin Lymphoma. Clin. Transl. Oncol. 2020, 22, 782–785. [Google Scholar] [CrossRef]
- Gholiha, A.R.; Hollander, P.; Löf, L.; Glimelius, I.; Hedstrom, G.; Molin, D.; Hjalgrim, H.; Smedby, K.E.; Hashemi, J.; Amini, R.-M.; et al. Checkpoint CD47 Expression in Classical Hodgkin Lymphoma. Br. J. Haematol. 2022, 1–10. [Google Scholar] [CrossRef]
- Ansell, S.M.; Maris, M.B.; Lesokhin, A.M.; Chen, R.W.; Flinn, I.W.; Sawas, A.; Minden, M.D.; Villa, D.; Percival, M.-E.M.; Advani, A.S.; et al. Phase I Study of the CD47 Blocker TTI-621 in Patients with Relapsed or Refractory Hematologic Malignancies. Clin. Cancer Res. 2021, 27, 2190–2199. [Google Scholar] [CrossRef]
- Opinto, G.; Agostinelli, C.; Ciavarella, S.; Guarini, A.; Maiorano, E.; Ingravallo, G. Hodgkin Lymphoma: A Special Microenvironment. J. Clin. Med. 2021, 10, 4665. [Google Scholar] [CrossRef] [PubMed]
- Luminari, S.; Donati, B.; Casali, M.; Valli, R.; Santi, R.; Puccini, B.; Kovalchuk, S.; Ruffini, A.; Fama, A.; Berti, V.; et al. A Gene Expression–Based Model to Predict Metabolic Response After Two Courses of ABVD in Hodgkin Lymphoma Patients. Clin. Cancer Res. 2020, 26, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, R.L.; Mottok, A.; Chan, F.C.; Jiang, A.; Diepstra, A.; Visser, L.; Telenius, A.; Gascoyne, R.D.; Friedman, D.L.; Schwartz, C.L.; et al. A Gene Expression-Based Model Predicts Outcome in Children with Intermediate-Risk Classical Hodgkin Lymphoma. Blood 2022, 139, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Jachimowicz, R.D.; Klapper, W.; Glehr, G.; Müller, H.; Haverkamp, H.; Thorns, C.; Hansmann, M.L.; Möller, P.; Stein, H.; Rehberg, T.; et al. Gene Expression-Based Outcome Prediction in Advanced Stage Classical Hodgkin Lymphoma Treated with BEACOPP. Leukemia 2021, 35, 3589–3593. [Google Scholar] [CrossRef]
- Bertuzzi, C.; Sabattini, E.; Agostinelli, C. Immune Microenvironment Features and Dynamics in Hodgkin Lymphoma. Cancers 2021, 13, 3634. [Google Scholar] [CrossRef]
- Cader, F.Z.; Schackmann, R.C.J.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; et al. Mass Cytometry of Hodgkin Lymphoma Reveals a CD4+ Regulatory T-Cell-Rich and Exhausted T-Effector Microenvironment. Blood 2018, 132, 825–836. [Google Scholar] [CrossRef]
- Aldinucci, D.; Gloghini, A.; Pinto, A.; De Filippi, R.; Carbone, A. The Classical Hodgkin’s Lymphoma Microenvironment and Its Role in Promoting Tumour Growth and Immune Escape. J. Pathol. 2010, 221, 248–263. [Google Scholar] [CrossRef]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T Lymphocytes Coexpressing CCR4 and a Chimeric Antigen Receptor Targeting CD30 Have Improved Homing and Antitumor Activity in a Hodgkin Tumor Model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [Green Version]
- Tanijiri, T.; Shimizu, T.; Uehira, K.; Yokoi, T.; Amuro, H.; Sugimoto, H.; Torii, Y.; Tajima, K.; Ito, T.; Amakawa, R.; et al. Hodgkin’s Reed-Sternberg Cell Line (KM-H2) Promotes a Bidirectional Differentiation of CD4+CD25+Foxp3+ T Cells and CD4+ Cytotoxic T Lymphocytes from CD4+ Naive T Cells. J. Leukoc. Biol. 2007, 82, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Casagrande, N.; Borghese, C.; Visser, L.; Mongiat, M.; Colombatti, A.; Aldinucci, D. CCR5 Antagonism by Maraviroc Inhibits Hodgkin Lymphoma Microenvironment Interactions and Xenograft Growth. Haematologica 2019, 104, 564–575. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Lorenzon, D.; Cattaruzza, L.; Pinto, A.; Gloghini, A.; Carbone, A.; Colombatti, A. Expression of CCR5 Receptors on Reed-Sternberg Cells and Hodgkin Lymphoma Cell Lines: Involvement of CCL5/Rantes in Tumor Cell Growth and Microenvironmental Interactions. Int. J. Cancer 2008, 122, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Jalali, S.; Price-Troska, T.; Bothun, C.; Villasboas, J.; Kim, H.-J.; Yang, Z.-Z.; Novak, A.J.; Dong, H.; Ansell, S.M. Reverse Signaling via PD-L1 Supports Malignant Cell Growth and Survival in Classical Hodgkin Lymphoma. Blood Cancer J. 2019, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Rengstl, B.; Schmid, F.; Weiser, C.; Döring, C.; Heinrich, T.; Warner, K.; Becker, P.S.A.; Wistinghausen, R.; Kameh-Var, S.; Werling, E.; et al. Tumor-Infiltrating HLA-Matched CD4+ T Cells Retargeted against Hodgkin and Reed–Sternberg Cells. OncoImmunology 2016, 5, e1160186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacchiana, R.; Abbate, M.; Armato, U.; Dal Prà, I.; Chiarini, A. Combining Immunofluorescence with in Situ Proximity Ligation Assay: A Novel Imaging Approach to Monitor Protein-Protein Interactions in Relation to Subcellular Localization. Histochem. Cell Biol. 2014, 142, 593–600. [Google Scholar] [CrossRef]
- Zocchi, M.R.; Catellani, S.; Canevali, P.; Tavella, S.; Garuti, A.; Villaggio, B.; Zunino, A.; Gobbi, M.; Fraternali-Orcioni, G.; Kunkl, A.; et al. High ERp5/ADAM10 Expression in Lymph Node Microenvironment and Impaired NKG2D Ligands Recognition in Hodgkin Lymphomas. Blood 2012, 119, 1479–1489. [Google Scholar] [CrossRef]
- Fischer, M.; Juremalm, M.; Olsson, N.; Backlin, C.; Sundström, C.; Nilsson, K.; Enblad, G.; Nilsson, G. Expression of CCL5/RANTES by Hodgkin and Reed-Sternberg Cells and Its Possible Role in the Recruitment of Mast Cells into Lymphomatous Tissue. Int. J. Cancer 2003, 107, 197–201. [Google Scholar] [CrossRef]
- Pinto, A.; Aldinucci, D.; Gloghini, A.; Zagonel, V.; Degan, M.; Improta, S.; Juzbasic, S.; Todesco, M.; Perin, V.; Gattei, V.; et al. Human Eosinophils Express Functional CD30 Ligand and Stimulate Proliferation of a Hodgkin’s Disease Cell Line. Blood 1996, 88, 3299–3305. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.; Aldinucci, D.; Gloghini, A.; Zagonel, V.; Degan, M.; Perin, V.; Todesco, M.; De Iuliis, A.; Improta, S.; Sacco, C.; et al. The Role of Eosinophils in the Pathobiology of Hodgkin’s Disease. Ann. Oncol. 1997, 8 (Suppl. 2), 89–96. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 3151. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Bosshart, H.; Heinzelmann, M. THP-1 Cells as a Model for Human Monocytes. Ann. Transl. Med. 2016, 4, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tudor, C.S.; Bruns, H.; Daniel, C.; Distel, L.V.; Hartmann, A.; Gerbitz, A.; Buettner, M.J. Macrophages and Dendritic Cells as Actors in the Immune Reaction of Classical Hodgkin Lymphoma. PLoS ONE 2014, 9, e114345. [Google Scholar] [CrossRef] [Green Version]
- Ruella, M.; Klichinsky, M.; Kenderian, S.S.; Shestova, O.; Ziober, A.; Kraft, D.O.; Feldman, M.; Wasik, M.A.; June, C.H.; Gill, S. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. Cancer Discov. 2017, 7, 1154–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, M.; Carreras, J.; Higuchi, H.; Kotaki, R.; Hoshina, T.; Okuyama, K.; Suzuki, N.; Kakizaki, M.; Miyatake, Y.; Ando, K.; et al. PD-L1/L2 Protein Levels Rapidly Increase on Monocytes via Trogocytosis from Tumor Cells in Classical Hodgkin Lymphoma. Leukemia 2020, 34, 2405–2417. [Google Scholar] [CrossRef] [PubMed]
- Madsen, D.H.; Leonard, D.; Masedunskas, A.; Moyer, A.; Jürgensen, H.J.; Peters, D.E.; Amornphimoltham, P.; Selvaraj, A.; Yamada, S.S.; Brenner, D.A.; et al. M2-like Macrophages Are Responsible for Collagen Degradation through a Mannose Receptor–Mediated Pathway. J. Cell Biol. 2013, 202, 951–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, L.; Dreyer, J.H.; Hartmann, D.; Barros, M.H.M.; Büttner-Herold, M.; Grittner, U.; Niedobitek, G. Tumor-Associated Macrophages in Classical Hodgkin Lymphoma: Hormetic Relationship to Outcome. Sci. Rep. 2020, 10, 9410. [Google Scholar] [CrossRef]
- Cucchi, D.G.J.; Groen, R.W.J.; Janssen, J.J.W.M.; Cloos, J. Ex Vivo Cultures and Drug Testing of Primary Acute Myeloid Leukemia Samples: Current Techniques and Implications for Experimental Design and Outcome. Drug Resist. Updates 2020, 53, 100730. [Google Scholar] [CrossRef]
- Meadows, S.A.; Vega, F.; Kashishian, A.; Johnson, D.; Diehl, V.; Miller, L.L.; Younes, A.; Lannutti, B.J. PI3Kδ Inhibitor, GS-1101 (CAL-101), Attenuates Pathway Signaling, Induces Apoptosis, and Overcomes Signals from the Microenvironment in Cellular Models of Hodgkin Lymphoma. Blood 2012, 119, 1897–1900. [Google Scholar] [CrossRef]
- Dörsam, B.; Bösl, T.; Reiners, K.S.; Barnert, S.; Schubert, R.; Shatnyeva, O.; Zigrino, P.; Engert, A.; Hansen, H.P.; von Strandmann, E.P. Hodgkin Lymphoma-Derived Extracellular Vesicles Change the Secretome of Fibroblasts Toward a CAF Phenotype. Front. Immunol. 2018, 9, 1358. [Google Scholar] [CrossRef]
- Celegato, M.; Borghese, C.; Casagrande, N.; Mongiat, M.; Kahle, X.U.; Paulitti, A.; Spina, M.; Colombatti, A.; Aldinucci, D. Preclinical Activity of the Repurposed Drug Auranofin in Classical Hodgkin Lymphoma. Blood 2015, 126, 1394–1397. [Google Scholar] [CrossRef] [Green Version]
- Cattaruzza, L.; Gloghini, A.; Olivo, K.; Di Francia, R.; Lorenzon, D.; De Filippi, R.; Carbone, A.; Colombatti, A.; Pinto, A.; Aldinucci, D. Functional Coexpression of Interleukin (IL)-7 and Its Receptor (IL-7R) on Hodgkin and Reed-Sternberg Cells: Involvement of IL-7 in Tumor Cell Growth and Microenvironmental Interactions of Hodgkin’s Lymphoma. Int. J. Cancer 2009, 125, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Celegato, M.; Borghese, C.; Casagrande, N.; Carbone, A.; Colombatti, A.; Aldinucci, D. Bortezomib Down-Modulates the Survival Factor Interferon Regulatory Factor 4 in Hodgkin Lymphoma Cell Lines and Decreases the Protective Activity of Hodgkin Lymphoma-Associated Fibroblasts. Leuk. Lymphoma 2014, 55, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Celegato, M.; Borghese, C.; Umezawa, K.; Casagrande, N.; Colombatti, A.; Carbone, A.; Aldinucci, D. The NF-ΚB Inhibitor DHMEQ Decreases Survival Factors, Overcomes the Protective Activity of Microenvironment and Synergizes with Chemotherapy Agents in Classical Hodgkin Lymphoma. Cancer Lett. 2014, 349, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.L.; Bustos, S.O.; Bhatt, D.; Chammas, R.; de Sousa Andrade, L. N Tumor-Derived Extracellular Vesicles: Modulation of Cellular Functional Dynamics in Tumor Microenvironment and Its Clinical Implications. Front. Cell Dev. Biol. 2021, 9, 737449. [Google Scholar] [CrossRef] [PubMed]
- Trino, S.; Lamorte, D.; Caivano, A.; De Luca, L.; Sgambato, A.; Laurenzana, I. Clinical Relevance of Extracellular Vesicles in Hematological Neoplasms: From Liquid Biopsy to Cell Biopsy. Leukemia 2021, 35, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Tosetti, F.; Venè, R.; Camodeca, C.; Nuti, E.; Rossello, A.; D’Arrigo, C.; Galante, D.; Ferrari, N.; Poggi, A.; Zocchi, M.R. Specific ADAM10 Inhibitors Localize in Exosome-like Vesicles Released by Hodgkin Lymphoma and Stromal Cells and Prevent Sheddase Activity Carried to Bystander Cells. Oncoimmunology 2018, 7, e1421889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zocchi, M.R.; Camodeca, C.; Nuti, E.; Rossello, A.; Venè, R.; Tosetti, F.; Dapino, I.; Costa, D.; Musso, A.; Poggi, A. ADAM10 New Selective Inhibitors Reduce NKG2D Ligand Release Sensitizing Hodgkin Lymphoma Cells to NKG2D-Mediated Killing. Oncoimmunology 2015, 5, e1123367. [Google Scholar] [CrossRef] [Green Version]
- Hansen, H.P.; Engels, H.-M.; Dams, M.; Paes Leme, A.F.; Pauletti, B.A.; Simhadri, V.L.; Dürkop, H.; Reiners, K.S.; Barnert, S.; Engert, A.; et al. Protrusion-Guided Extracellular Vesicles Mediate CD30 Trans-Signalling in the Microenvironment of Hodgkin’s Lymphoma. J. Pathol. 2014, 232, 405–414. [Google Scholar] [CrossRef]
- Drees, E.E.E.; Roemer, M.G.M.; Groenewegen, N.J.; Perez-Boza, J.; van Eijndhoven, M.A.J.; Prins, L.I.; Verkuijlen, S.A.W.M.; Tran, X.-M.; Driessen, J.; Zwezerijnen, G.J.C.; et al. Extracellular Vesicle MiRNA Predict FDG-PET Status in Patients with Classical Hodgkin Lymphoma. J. Extracell. Vesicles 2021, 10, e12121. [Google Scholar] [CrossRef]
- Repetto, O.; Lovisa, F.; Elia, C.; Enderle, D.; Romanato, F.; Buffardi, S.; Sala, A.; Pillon, M.; Steffan, A.; Burnelli, R.; et al. Proteomic Exploration of Plasma Exosomes and Other Small Extracellular Vesicles in Pediatric Hodgkin Lymphoma: A Potential Source of Biomarkers for Relapse Occurrence. Diagnostics 2021, 11, 917. [Google Scholar] [CrossRef]
- Slyusarenko, M.; Shalaev, S.; Valitova, A.; Zabegina, L.; Nikiforova, N.; Nazarova, I.; Rudakovskaya, P.; Vorobiev, M.; Lezov, A.; Filatova, L.; et al. AuNP Aptasensor for Hodgkin Lymphoma Monitoring. Biosensors 2022, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, M.; Higuchi, H.; Kotani, A. Significance of Trogocytosis and Exosome-Mediated Transport in Establishing and Maintaining the Tumor Microenvironment in Lymphoid Malignancies. J. Clin. Exp. Hematop. 2021, 61, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Schwarz, H. The Role of Trogocytosis in Immune Surveillance of Hodgkin Lymphoma. Oncoimmunology 2020, 9, 1781334. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, S.; Ho, W.T.; Schwarz, H. CD137 Signaling in Hodgkin and Reed-Sternberg Cell Lines Induces IL-13 Secretion, Immune Deviation and Enhanced Growth. Oncoimmunology 2016, 5, e1160188. [Google Scholar] [CrossRef] [Green Version]
- Pang, W.L.; Ho, W.T.; Schwarz, H. Ectopic CD137 Expression Facilitates the Escape of Hodgkin and Reed-Sternberg Cells from Immunosurveillance. Oncoimmunology 2013, 2, e23441. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.T.; Pang, W.L.; Chong, S.M.; Castella, A.; Al-Salam, S.; Tan, T.E.; Moh, M.C.; Koh, L.K.; Gan, S.U.; Cheng, C.K.; et al. Expression of CD137 on Hodgkin and Reed-Sternberg Cells Inhibits T-Cell Activation by Eliminating CD137 Ligand Expression. Cancer Res. 2013, 73, 652–661. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, M.; Watanabe, M.; Uchimaru, K.; Horie, R. Trogocytosis of Ligand-Receptor Complex and Its Intracellular Transport in CD30 Signalling. Biol. Cell 2018, 110, 109–124. [Google Scholar] [CrossRef]
- Li, Z.; Ju, X.; Lee, K.; Clarke, C.; Hsu, J.L.; Abadir, E.; Bryant, C.E.; Pears, S.; Sunderland, N.; Heffernan, S.; et al. CD83 Is a New Potential Biomarker and Therapeutic Target for Hodgkin Lymphoma. Haematologica 2018, 103, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Colombo, E.; Cattaneo, M.G. Multicellular 3D Models to Study Tumour-Stroma Interactions. Int. J. Mol. Sci. 2021, 22, 1633. [Google Scholar] [CrossRef]
- Farhat, J.; Pandey, I.; AlWahsh, M. Transcending toward Advanced 3D-Cell Culture Modalities: A Review about an Emerging Paradigm in Translational Oncology. Cells 2021, 10, 1657. [Google Scholar] [CrossRef]
- Ikram, M.; Lim, Y.; Baek, S.-Y.; Jin, S.; Jeong, Y.H.; Kwak, J.-Y.; Yoon, S. Co-Targeting of Tiam1/Rac1 and Notch Ameliorates Chemoresistance against Doxorubicin in a Biomimetic 3D Lymphoma Model. Oncotarget 2018, 9, 2058–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyhan, A.A. Lost in Translation: The Valley of Death across Preclinical and Clinical Divide – Identification of Problems and Overcoming Obstacles. Transl. Med. Commun. 2019, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef] [PubMed]
- Pece, R.; Tavella, S.; Costa, D.; Varesano, S.; Camodeca, C.; Cuffaro, D.; Nuti, E.; Rossello, A.; Alfano, M.; D’Arrigo, C.; et al. Inhibitors of A Disintegrin And Metalloproteinases-10 Reduce Hodgkin Lymphoma Cell Growth in 3D Microenvironments and Enhance Brentuximab-Vedotin Effect. Haematologica 2021, 107, 909–920. [Google Scholar] [CrossRef]
- Birgersdotter, A.; Baumforth, K.R.N.; Porwit, A.; Sundblad, A.; Falk, K.I.; Wei, W.; Sjöberg, J.; Murray, P.G.; Björkholm, M.; Ernberg, I. Three-Dimensional Culturing of the Hodgkin Lymphoma Cell-Line L1236 Induces a HL Tissue-like Gene Expression Pattern. Leuk. Lymphoma 2007, 48, 2042–2053. [Google Scholar] [CrossRef]
- Brix, N.; Samaga, D.; Belka, C.; Zitzelsberger, H.; Lauber, K. Analysis of Clonogenic Growth in vitro. Nat. Protoc. 2021, 16, 4963–4991. [Google Scholar] [CrossRef]
- Casagrande, N.; Borghese, C.; Aldinucci, D. In Classical Hodgkin Lymphoma the Combination of the CCR5 Antagonist Maraviroc with Trabectedin Synergizes, Enhances DNA Damage and Decreases Three-Dimensional Tumor-Stroma Heterospheroid Viability. Haematologica 2022, 107, 287–291. [Google Scholar] [CrossRef]
- Hansen, H.P.; Trad, A.; Dams, M.; Zigrino, P.; Moss, M.; Tator, M.; Schön, G.; Grenzi, P.C.; Bachurski, D.; Aquino, B.; et al. CD30 on Extracellular Vesicles from Malignant Hodgkin Cells Supports Damaging of CD30 Ligand-Expressing Bystander Cells with Brentuximab-Vedotin, in Vitro. Oncotarget 2016, 7, 30523–30535. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Poletto, D.; Gloghini, A.; Nanni, P.; Degan, M.; Perin, T.; Ceolin, P.; Rossi, F.M.; Gattei, V.; Carbone, A.; et al. Expression of Functional Interleukin-3 Receptors on Hodgkin and Reed-Sternberg Cells. Am. J. Pathol. 2002, 160, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.; Heinrich, M.A.; Teixeira, L.M.; Prakash, J. 3D In Vitro Model (R)Evolution: Unveiling Tumor–Stroma Interactions. Trends Cancer 2021, 7, 249–264. [Google Scholar] [CrossRef]
- Goncharova, O.; Flinner, N.; Bein, J.; Döring, C.; Donnadieu, E.; Rikirsch, S.; Herling, M.; Küppers, R.; Hansmann, M.-L.; Hartmann, S. Migration Properties Distinguish Tumor Cells of Classical Hodgkin Lymphoma from Anaplastic Large Cell Lymphoma Cells. Cancers 2019, 11, 1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fhu, C.W.; Graham, A.M.; Yap, C.T.; Al-Salam, S.; Castella, A.; Chong, S.M.; Lim, Y.-C. Reed-Sternberg Cell-Derived Lymphotoxin-α Activates Endothelial Cells to Enhance T-Cell Recruitment in Classical Hodgkin Lymphoma. Blood 2014, 124, 2973–2982. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Tamma, R.; Annese, T. Chorioallantoic Membrane Vascularization. A Meta-Analysis. Exp. Cell Res. 2021, 405, 112716. [Google Scholar] [CrossRef] [PubMed]
- Linke, F.; Zaunig, S.; Nietert, M.M.; von Bonin, F.; Lutz, S.; Dullin, C.; Janovská, P.; Beissbarth, T.; Alves, F.; Klapper, W.; et al. WNT5A: A Motility-Promoting Factor in Hodgkin Lymphoma. Oncogene 2017, 36, 13–23. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casagrande, N.; Borghese, C.; Aldinucci, D. Current and Emerging Approaches to Study Microenvironmental Interactions and Drug Activity in Classical Hodgkin Lymphoma. Cancers 2022, 14, 2427. https://doi.org/10.3390/cancers14102427
Casagrande N, Borghese C, Aldinucci D. Current and Emerging Approaches to Study Microenvironmental Interactions and Drug Activity in Classical Hodgkin Lymphoma. Cancers. 2022; 14(10):2427. https://doi.org/10.3390/cancers14102427
Chicago/Turabian StyleCasagrande, Naike, Cinzia Borghese, and Donatella Aldinucci. 2022. "Current and Emerging Approaches to Study Microenvironmental Interactions and Drug Activity in Classical Hodgkin Lymphoma" Cancers 14, no. 10: 2427. https://doi.org/10.3390/cancers14102427