
p62 Promotes Survival and Hepatocarcinogenesis in Mice with Liver-Specific NEMO Ablation

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
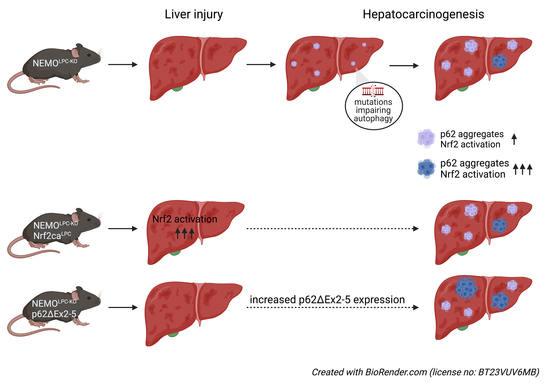
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Hepatocyte Isolation and Culture
2.3. Biochemical Serum Analyses
2.4. Antibodies
2.5. Immunoblotting
2.6. Immunofluorescence (IF) on Liver Sections and Primary Hepatocytes
2.7. Histology and Immunohistochemistry (IHC)
2.8. Image Quantification
2.9. Quantification of Macroscopically Visible Tumors and Histopathological Evaluation
2.10. Electron Microscopy
2.11. Quantitative RT–PCR
2.12. Statistical Analysis
3. Results
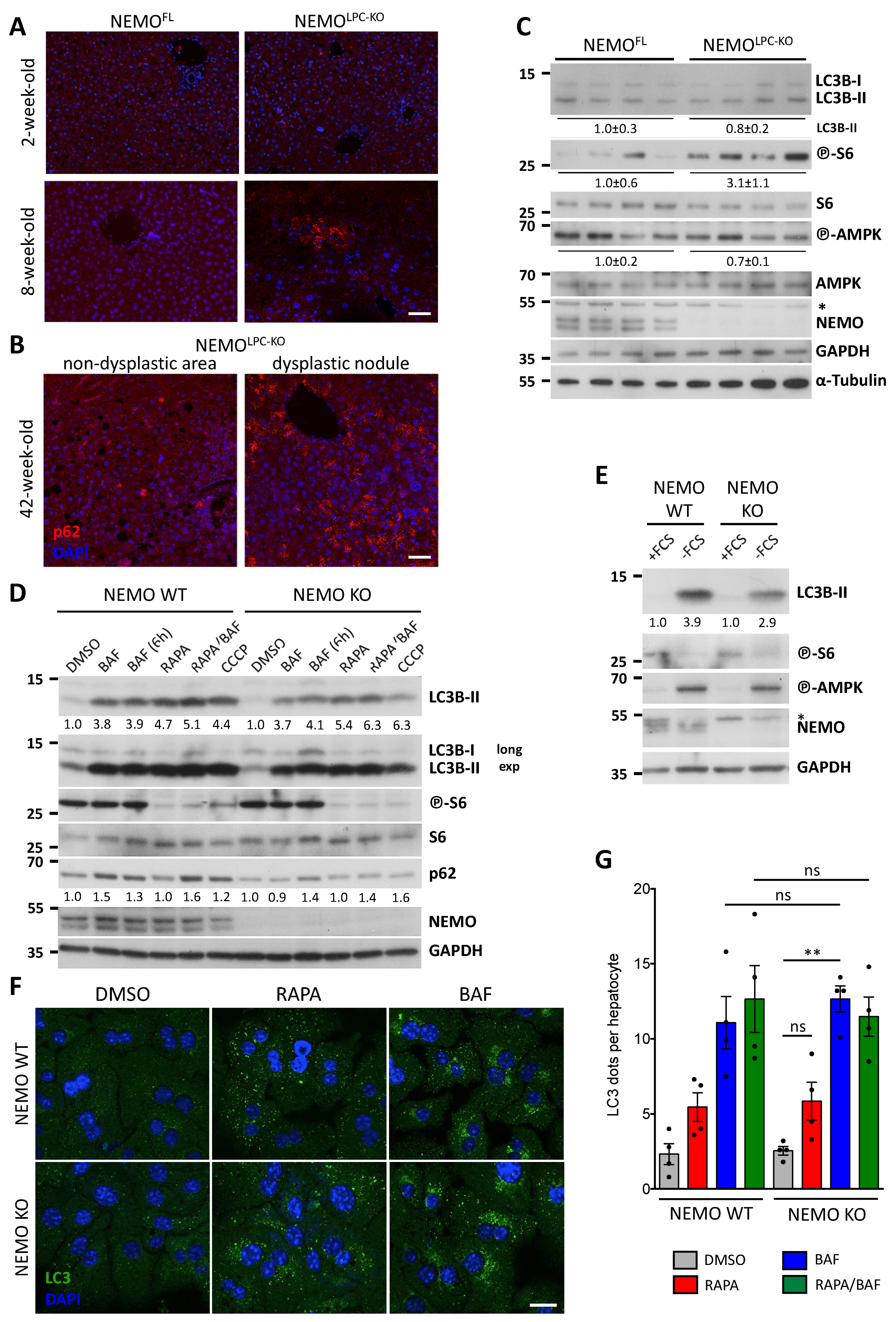
3.1. Focal p62 Accumulation and mTOR Activation in Hepatocytes of NEMOLPC-KO Mice without Autophagy Inhibition
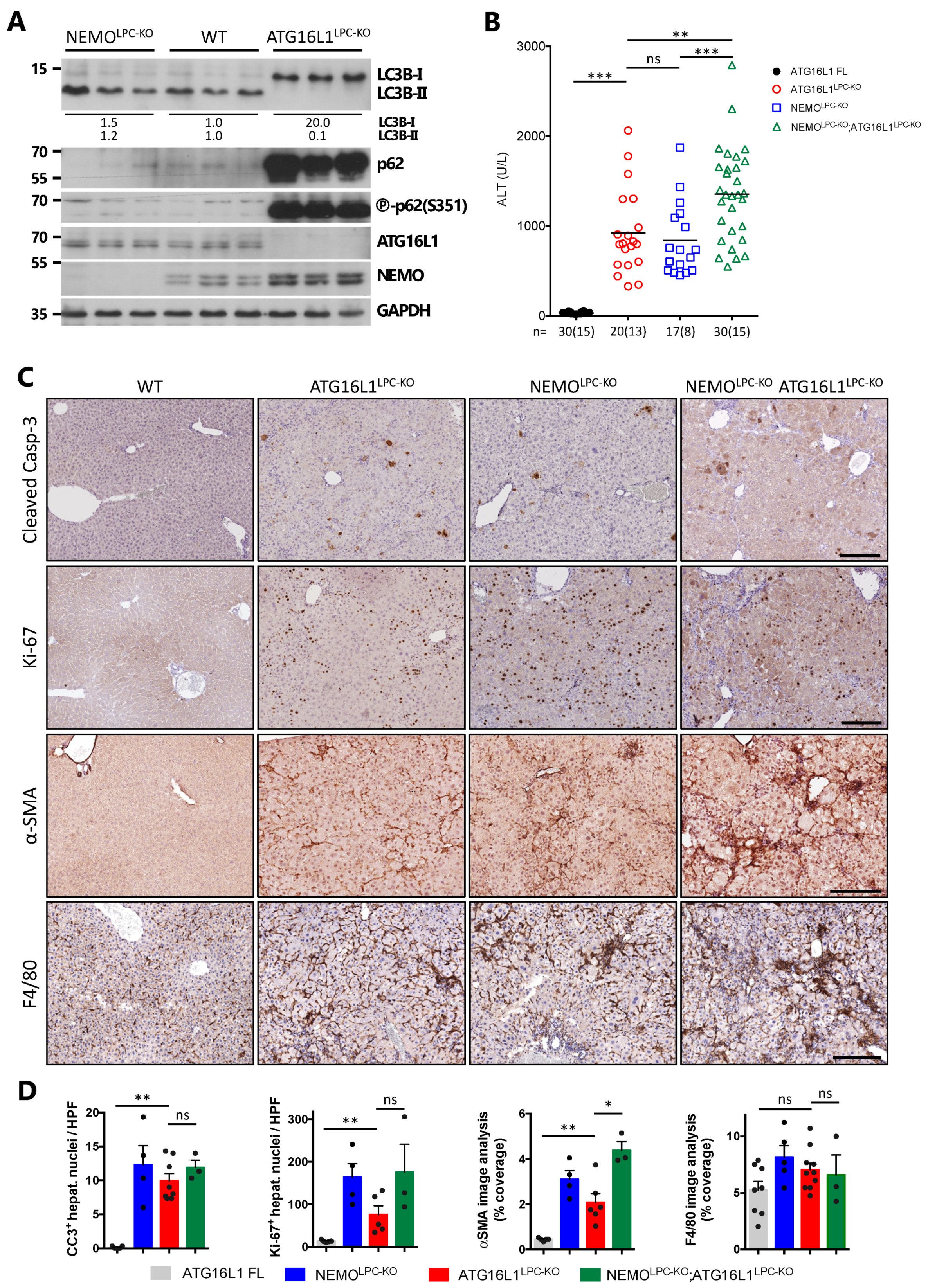
3.2. Hepatic NEMO and Autophagy Protect from CLD via Distinct Mechanisms
3.3. p62 Deficiency Impairs Survival of NEMOLPC-KO Mice
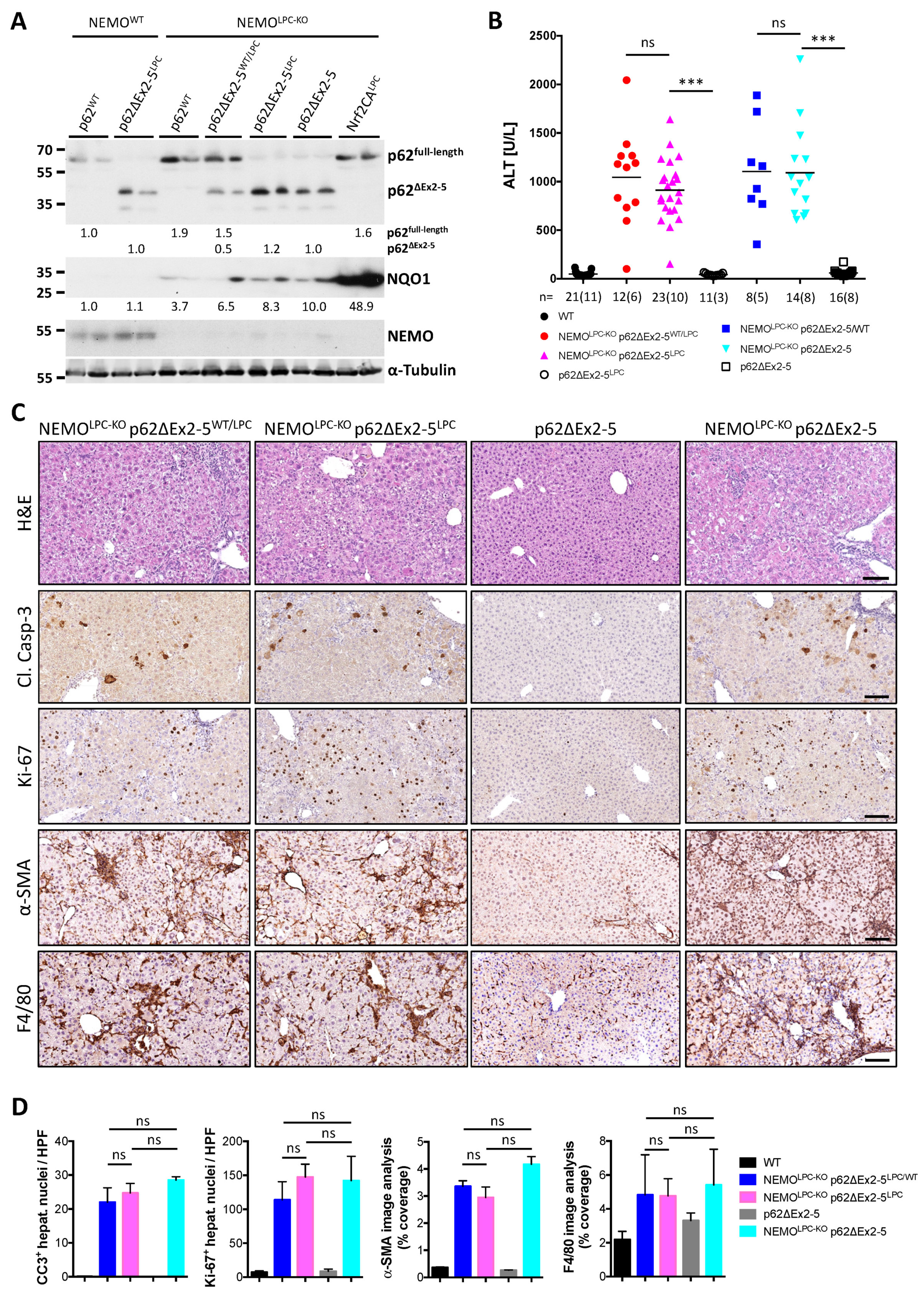
3.4. Expression of a p62 Mutant Aggravates Hepatocarcinogenesis in NEMOLPC-KO Mice
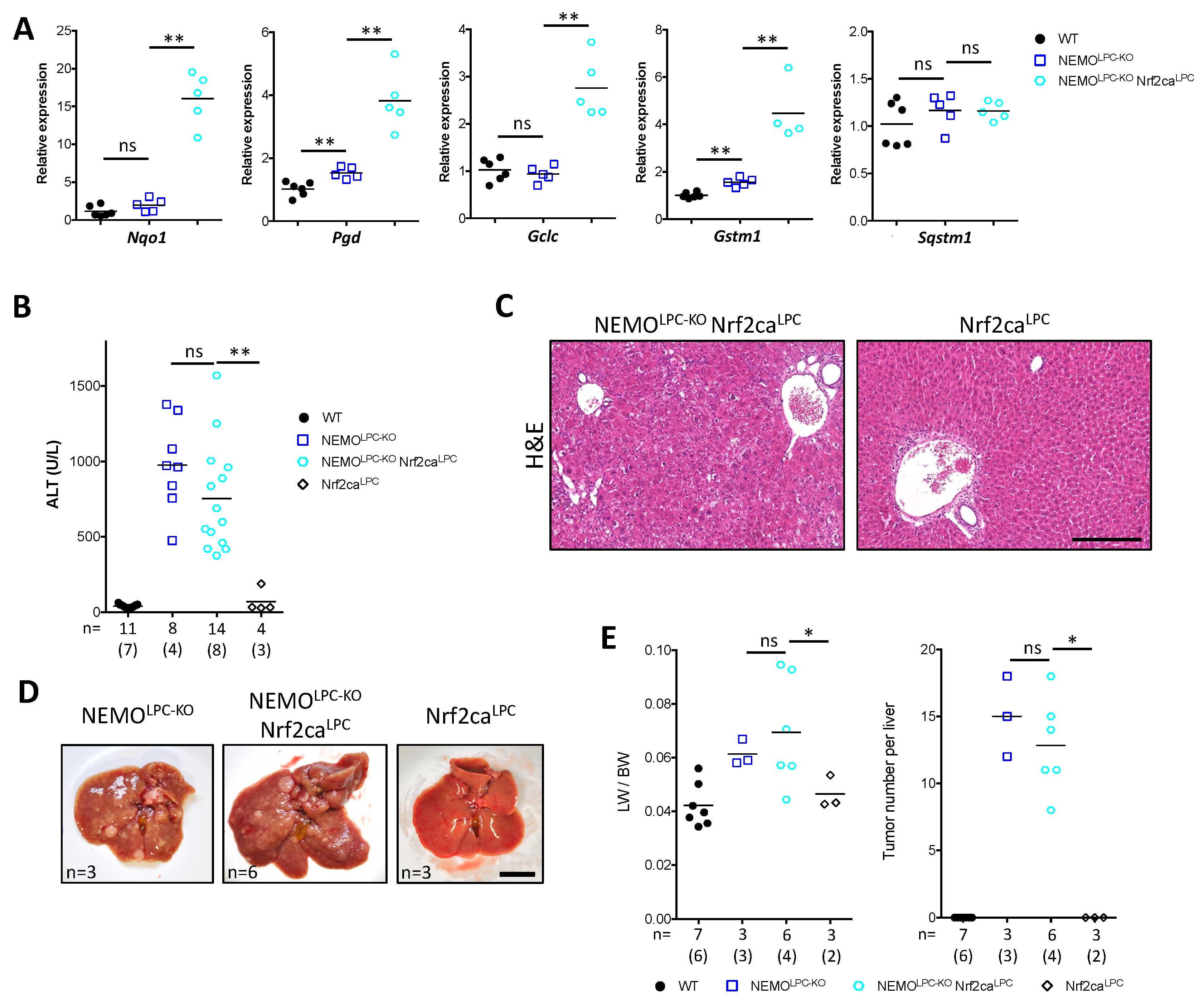
3.5. Nrf2 Overactivation Per Se Does Not Affect Liver Damage and Tumorigenesis in NEMOLPC-KO Mice
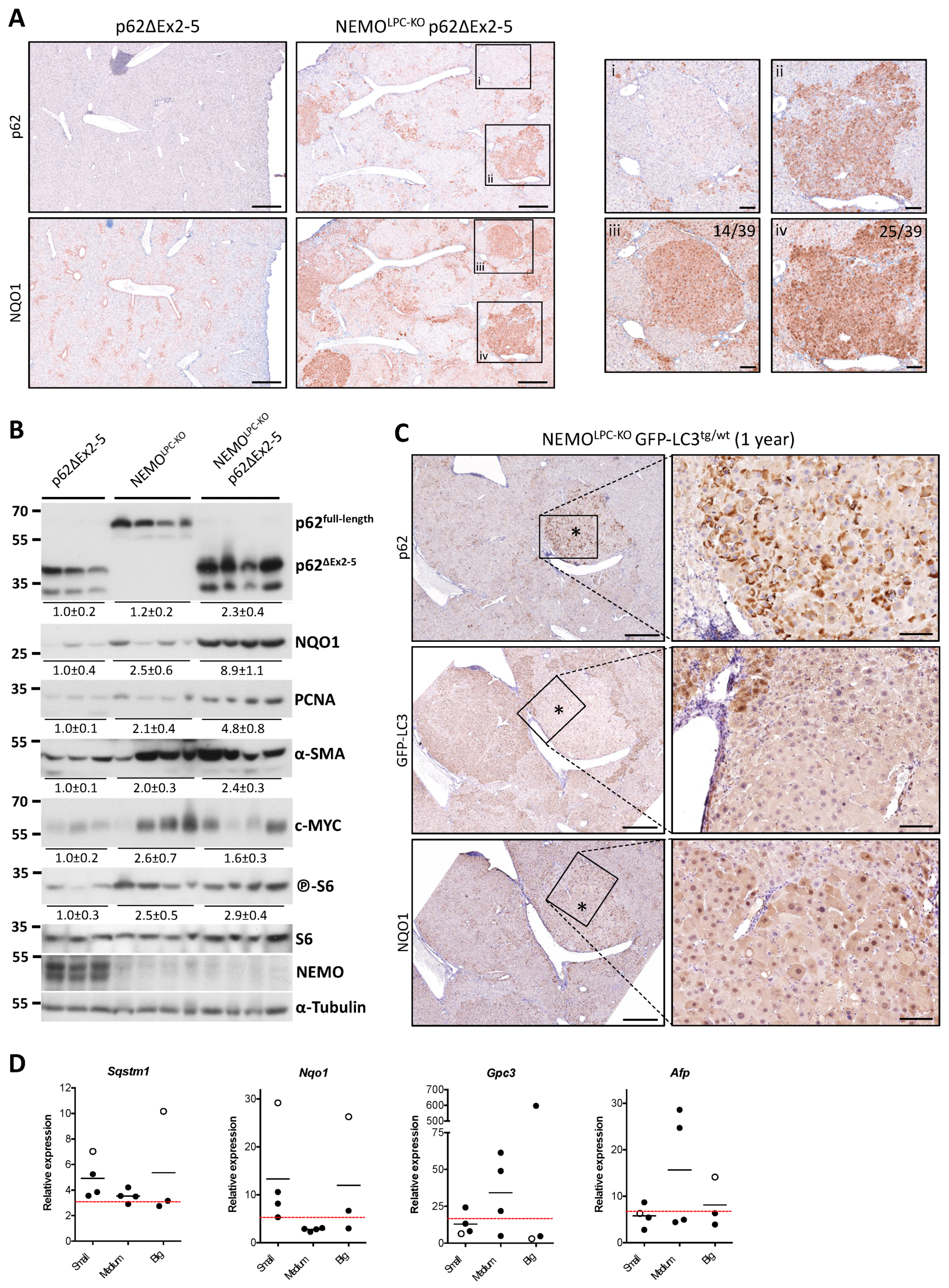
3.6. Aberrant p62 Accumulation and Acquired Autophagy Defects Associate with p62-Keap1-Nrf2 Positive Liver Tumors
4. Discussion
4.1. IKK Complex and Autophagy Independently Regulate Liver Homeostasis
4.2. The Role of p62 and Nrf2 in Liver Injury Is Context-Dependent
4.3. Defective Autophagy Is Required to Potentiate the Hepatocarcinogenic Role of the p62-Keap1-Nrf2 Axis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guicciardi, M.E.; Malhi, H.; Mott, J.L.; Gores, G.J. Apoptosis and necrosis in the liver. Compr. Physiol. 2013, 3, 977–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, R.F.; Luedde, T. Apoptosis and necroptosis in the liver: A matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Kondylis, V.; Pasparakis, M. RIP Kinases in Liver Cell Death, Inflammation and Cancer. Trends Mol. Med. 2019, 25, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Kondylis, V.; Polykratis, A.; Ehlken, H.; Ochoa-Callejero, L.; Straub, B.K.; Krishna-Subramanian, S.; Van, T.M.; Curth, H.M.; Heise, N.; Weih, F.; et al. NEMO Prevents Steatohepatitis and Hepatocellular Carcinoma by Inhibiting RIPK1 Kinase Activity-Mediated Hepatocyte Apoptosis. Cancer Cell 2015, 28, 582–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef]
- Ting, A.T.; Bertrand, M.J.M. More to Life than NF-kappaB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Dondelinger, Y.; Jouan-Lanhouet, S.; Divert, T.; Theatre, E.; Bertin, J.; Gough, P.J.; Giansanti, P.; Heck, A.J.; Dejardin, E.; Vandenabeele, P.; et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 2015, 60, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Ehlken, H.; Krishna-Subramanian, S.; Ochoa-Callejero, L.; Kondylis, V.; Nadi, N.E.; Straub, B.K.; Schirmacher, P.; Walczak, H.; Kollias, G.; Pasparakis, M. Death receptor-independent FADD signalling triggers hepatitis and hepatocellular carcinoma in mice with liver parenchymal cell-specific NEMO knockout. Cell Death Differ. 2014, 21, 1721–1732. [Google Scholar] [CrossRef] [Green Version]
- Criollo, A.; Senovilla, L.; Authier, H.; Maiuri, M.C.; Morselli, E.; Vitale, I.; Kepp, O.; Tasdemir, E.; Galluzzi, L.; Shen, S.; et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010, 29, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Comb, W.C.; Cogswell, P.; Sitcheran, R.; Baldwin, A.S. IKK-dependent, NF-kappaB-independent control of autophagic gene expression. Oncogene 2011, 30, 1727–1732. [Google Scholar] [CrossRef] [Green Version]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, death, and autophagy in cancer: NF-kappaB turns up everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inokuchi-Shimizu, S.; Park, E.J.; Roh, Y.S.; Yang, L.; Zhang, B.; Song, J.; Liang, S.; Pimienta, M.; Taniguchi, K.; Wu, X.; et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J. Clin. Investig. 2014, 124, 3566–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero-Martin, G.; Hoyer-Hansen, M.; Garcia-Garcia, C.; Fumarola, C.; Farkas, T.; Lopez-Rivas, A.; Jaattela, M. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009, 28, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Wu, X.; Holzer, R.G.; Lee, J.H.; Todoric, J.; Park, E.J.; Ogata, H.; Gukovskaya, A.S.; Gukovsky, I.; Pizzo, D.P.; et al. Loss of acinar cell IKKalpha triggers spontaneous pancreatitis in mice. J. Clin. Investig. 2013, 123, 2231–2243. [Google Scholar] [CrossRef] [Green Version]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [Green Version]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strnad, P.; Stumptner, C.; Zatloukal, K.; Denk, H. Intermediate filament cytoskeleton of the liver in health and disease. Histochem. Cell Biol. 2008, 129, 735–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martin, P.; Komatsu, M. p62/SQSTM1—Steering the cell through health and disease. J. Cell Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [Green Version]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Shibata, T.; Aburatani, H. Exploration of liver cancer genomes. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 340–349. [Google Scholar] [CrossRef]
- Orru, C.; Giordano, S.; Columbano, A. Nrf2 in Neoplastic and Non-Neoplastic Liver Diseases. Cancers 2020, 12, 2932. [Google Scholar] [CrossRef]
- Schmidt-Supprian, M.; Bloch, W.; Courtois, G.; Addicks, K.; Israel, A.; Rajewsky, K.; Pasparakis, M. NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol. Cell 2000, 5, 981–992. [Google Scholar] [CrossRef]
- Pasparakis, M.; Courtois, G.; Hafner, M.; Schmidt-Supprian, M.; Nenci, A.; Toksoy, A.; Krampert, M.; Goebeler, M.; Gillitzer, R.; Israel, A.; et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature 2002, 417, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Gareus, R.; Huth, M.; Breiden, B.; Nenci, A.; Rosch, N.; Haase, I.; Bloch, W.; Sandhoff, K.; Pasparakis, M. Normal epidermal differentiation but impaired skin-barrier formation upon keratinocyte-restricted IKK1 ablation. Nat. Cell Biol. 2007, 9, 461–469. [Google Scholar] [CrossRef]
- Sasaki, Y.; Derudder, E.; Hobeika, E.; Pelanda, R.; Reth, M.; Rajewsky, K.; Schmidt-Supprian, M. Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity 2006, 24, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Bock, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, A.; Duran, A.; Selloum, M.; Champy, M.F.; Diez-Guerra, F.J.; Flores, J.M.; Serrano, M.; Auwerx, J.; Diaz-Meco, M.T.; Moscat, J. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006, 3, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Skarnes, W.C.; Rosen, B.; West, A.P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A.O.; Thomas, M.; Harrow, J.; Cox, T.; et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011, 474, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef]
- Schafer, M.; Farwanah, H.; Willrodt, A.H.; Huebner, A.J.; Sandhoff, K.; Roop, D.; Hohl, D.; Bloch, W.; Werner, S. Nrf2 links epidermal barrier function with antioxidant defense. EMBO Mol. Med. 2012, 4, 364–379. [Google Scholar] [CrossRef]
- Kellendonk, C.; Opherk, C.; Anlag, K.; Schutz, G.; Tronche, F. Hepatocyte-specific expression of Cre recombinase. Genesis 2000, 26, 151–153. [Google Scholar] [CrossRef]
- Ehlken, H.; Kondylis, V.; Heinrichsdorff, J.; Ochoa-Callejero, L.; Roskams, T.; Pasparakis, M. Hepatocyte IKK2 protects Mdr2-/- mice from chronic liver failure. PLoS ONE 2011, 6, e25942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Yecies, J.L.; Zhang, H.H.; Howell, J.J.; Nicholatos, J.; Harputlugil, E.; Bronson, R.T.; Kwiatkowski, D.J.; Manning, B.D. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci. Signal. 2012, 5, ra24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Krishna-Subramanian, S.; Singer, S.; Armaka, M.; Banales, J.M.; Holzer, K.; Schirmacher, P.; Walczak, H.; Kollias, G.; Pasparakis, M.; Kondylis, V. RIPK1 and death receptor signaling drive biliary damage and early liver tumorigenesis in mice with chronic hepatobiliary injury. Cell Death Differ. 2019, 26, 2710–2726. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Su, Z.Y.; Kong, A.N. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [Green Version]
- Kohler, U.A.; Kurinna, S.; Schwitter, D.; Marti, A.; Schafer, M.; Hellerbrand, C.; Speicher, T.; Werner, S. Activated Nrf2 impairs liver regeneration in mice by activation of genes involved in cell-cycle control and apoptosis. Hepatology 2014, 60, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell 2011, 44, 134–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, A.; Hernandez, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Zhang, H.; Zheng, J.; Liu, Y. Glypican-3: A New Target for Diagnosis and Treatment of Hepatocellular Carcinoma. J. Cancer 2020, 11, 2008–2021. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Luedde, T.; Heinrichsdorff, J.; de Lorenzi, R.; De Vos, R.; Roskams, T.; Pasparakis, M. IKK1 and IKK2 cooperate to maintain bile duct integrity in the liver. Proc. Natl. Acad. Sci. USA 2008, 105, 9733–9738. [Google Scholar] [CrossRef] [Green Version]
- Criollo, A.; Niso-Santano, M.; Malik, S.A.; Michaud, M.; Morselli, E.; Marino, G.; Lachkar, S.; Arkhipenko, A.V.; Harper, F.; Pierron, G.; et al. Inhibition of autophagy by TAB2 and TAB3. EMBO J. 2011, 30, 4908–4920. [Google Scholar] [CrossRef]
- Tusco, R.; Jacomin, A.C.; Jain, A.; Penman, B.S.; Larsen, K.B.; Johansen, T.; Nezis, I.P. Kenny mediates selective autophagic degradation of the IKK complex to control innate immune responses. Nat. Commun. 2017, 8, 1264. [Google Scholar] [CrossRef]
- Yang, S.; Qiang, L.; Sample, A.; Shah, P.; He, Y.Y. NF-kappaB Signaling Activation Induced by Chloroquine Requires Autophagosome, p62 Protein, and c-Jun N-terminal Kinase (JNK) Signaling and Promotes Tumor Cell Resistance. J. Biol. Chem. 2017, 292, 3379–3388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, X.; Wang, S.; Fulte, S.; Ma, X.; Ahamed, F.; Cui, W.; Liu, Z.; Rulicke, T.; Zatloukal, K.; Zong, W.X.; et al. Hepatocytic p62 suppresses ductular reaction and tumorigenesis in mouse livers with mTORC1 activation and defective autophagy. J. Hepatol. 2022, 76, 639–651. [Google Scholar] [CrossRef]
- Kageyama, S.; Gudmundsson, S.R.; Sou, Y.S.; Ichimura, Y.; Tamura, N.; Kazuno, S.; Ueno, T.; Miura, Y.; Noshiro, D.; Abe, M.; et al. p62/SQSTM1-droplet serves as a platform for autophagosome formation and anti-oxidative stress response. Nat. Commun. 2021, 12, 16. [Google Scholar] [CrossRef]
- Delanghe, T.; Huyghe, J.; Lee, S.; Priem, D.; Van Coillie, S.; Gilbert, B.; Choi, S.M.; Vandenabeele, P.; Degterev, A.; Cuny, G.D.; et al. Antioxidant and food additive BHA prevents TNF cytotoxicity by acting as a direct RIPK1 inhibitor. Cell Death Dis. 2021, 12, 699. [Google Scholar] [CrossRef]
- Orru, C.; Perra, A.; Kowalik, M.A.; Rizzolio, S.; Puliga, E.; Cabras, L.; Giordano, S.; Columbano, A. Distinct Mechanisms Are Responsible for Nrf2-Keap1 Pathway Activation at Different Stages of Rat Hepatocarcinogenesis. Cancers 2020, 12, 2305. [Google Scholar] [CrossRef]
- Denk, H.; Stumptner, C.; Fuchsbichler, A.; Muller, T.; Farr, G.; Muller, W.; Terracciano, L.; Zatloukal, K. Are the Mallory bodies and intracellular hyaline bodies in neoplastic and non-neoplastic hepatocytes related? J. Pathol. 2006, 208, 653–661. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody Name | Supplier | Product Number |
|---|---|---|
| p62 | Progen | Cat#GP62-C |
| p-p62(S351) | MBL | Cat#PM074 |
| GAPDH | Novus Biologicals | Cat#NB300-221 |
| LC3B | Nanotools | Cat# 0231-100/LC3-5F10 |
| LC3B | Novusbio | Cat#NB600-1384 |
| p-S6 | Cell Signaling | Cat#5364 |
| S6 | Cell Signaling | Cat#2217 |
| p-S6K | Cell Signaling | Cat#9205 |
| p-AMPK | Cell Signaling | Cat#2535 |
| AMPK | Cell Signaling | Cat#2532 |
| α-Tubulin | Sigma- Aldrich | Cat#T6074 |
| Actin | Santa Cruz | Cat#sc-1616 |
| NEMO | Homemade | [4] |
| IKK2 | Imgenex | Cat#IMG-129A |
| ATG16L | MBL | Cat#M150-3 |
| Cleaved Casp-3 | Cell Signaling | Cat#9661 |
| Ki-67 | DAKO | Cat#M724901 |
| PCNA | Santa Cruz | Cat#sc-56 |
| α-SMA | Sigma-Aldrich | Cat#A2547 |
| F4/80 | Bio-Rad | Cat#MCA497GA |
| Ubiquitin | Enzo | Cat#PW8810/0500 |
| FADD | Upstate | Cat#05-486 |
| GFP | Abcam | Cat#Ab290 |
| Nqo1 | Elabscience | Cat#E-AB-16011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondylis, V.; Schneider, F.; Schorn, F.; Oikonomou, N.; Straub, B.K.; Werner, S.; Rosenstiel, P.; Pasparakis, M. p62 Promotes Survival and Hepatocarcinogenesis in Mice with Liver-Specific NEMO Ablation. Cancers 2022, 14, 2436. https://doi.org/10.3390/cancers14102436
Kondylis V, Schneider F, Schorn F, Oikonomou N, Straub BK, Werner S, Rosenstiel P, Pasparakis M. p62 Promotes Survival and Hepatocarcinogenesis in Mice with Liver-Specific NEMO Ablation. Cancers. 2022; 14(10):2436. https://doi.org/10.3390/cancers14102436
Chicago/Turabian StyleKondylis, Vangelis, Farina Schneider, Fabian Schorn, Nikos Oikonomou, Beate Katharina Straub, Sabine Werner, Philip Rosenstiel, and Manolis Pasparakis. 2022. "p62 Promotes Survival and Hepatocarcinogenesis in Mice with Liver-Specific NEMO Ablation" Cancers 14, no. 10: 2436. https://doi.org/10.3390/cancers14102436
APA StyleKondylis, V., Schneider, F., Schorn, F., Oikonomou, N., Straub, B. K., Werner, S., Rosenstiel, P., & Pasparakis, M. (2022). p62 Promotes Survival and Hepatocarcinogenesis in Mice with Liver-Specific NEMO Ablation. Cancers, 14(10), 2436. https://doi.org/10.3390/cancers14102436