
Leveraging STING, Batf3 Dendritic Cells, CXCR3 Ligands, and Other Components Related to Innate Immunity to Induce a “Hot” Tumor Microenvironment That Is Responsive to Immunotherapy

Abstract
:Simple Summary
Abstract
1. Introduction
2. How Are Endogenous T Cells Successfully Recruited into The Tumor Microenvironment?
Clinical Applications
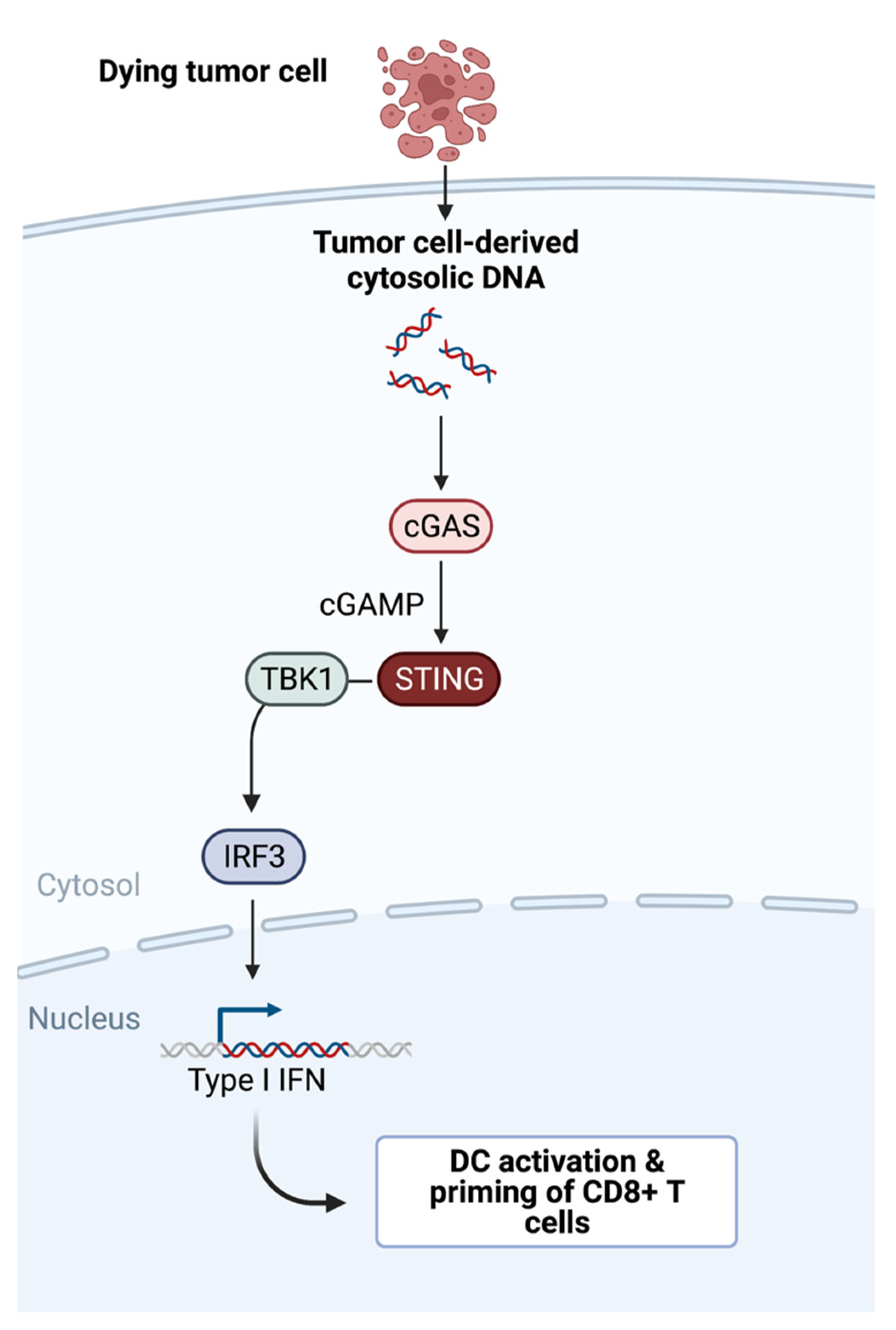
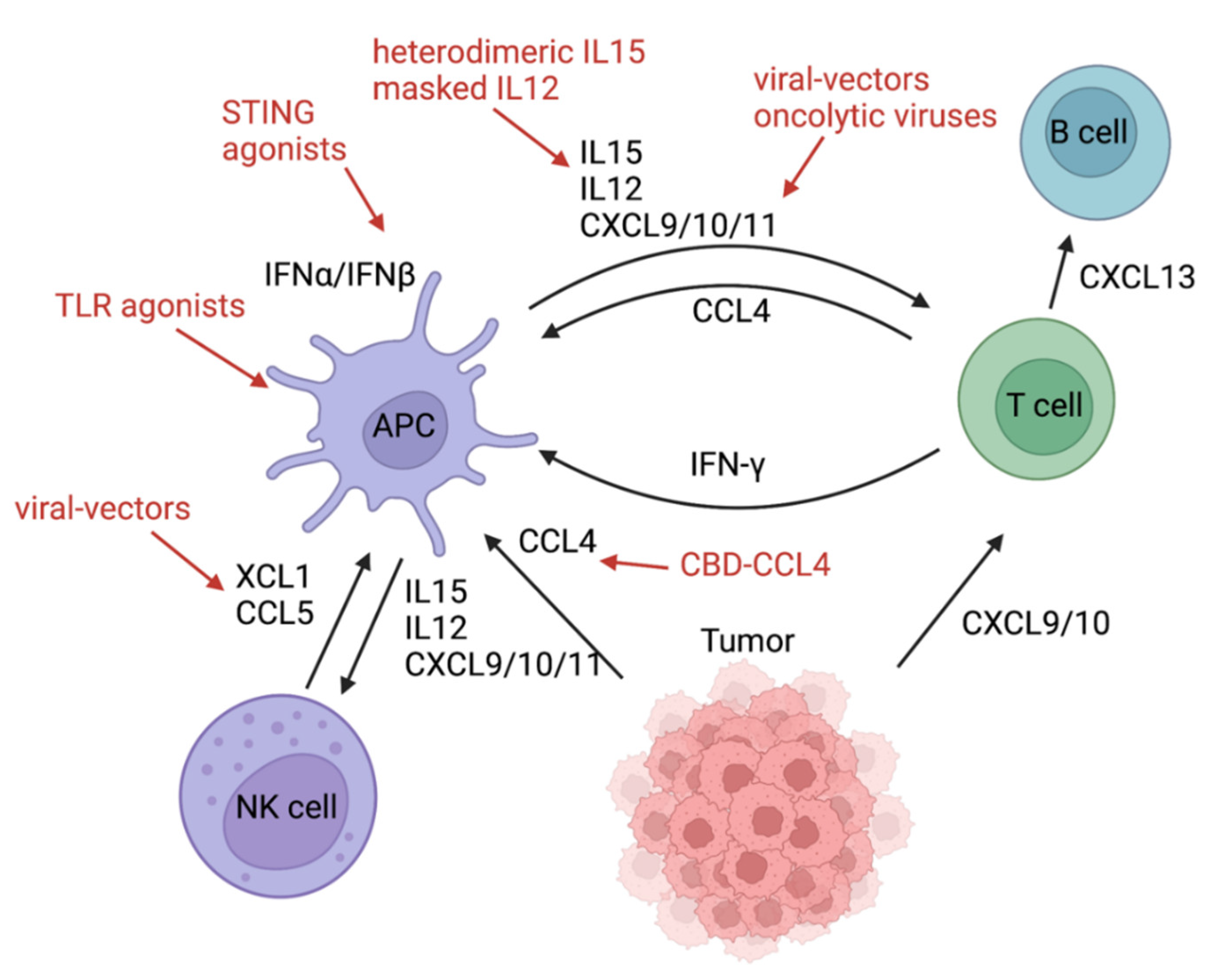
3. Batf3 DCs Initiating a Chemokine-Cytokine Network in the TME
3.1. Inducing Chemokines and Cytokines
3.2. Adverse Effects Caused by Chemokines and Cytokines

{kind=link}
{kind=link}
{kind=link}
| Components | Function | Species | Delivery Route/Therapeutic Agent | Year/Citation |
|---|---|---|---|---|
| STING | Sensing tumor-derived DNA, DC maturation, and CTL priming | mouse | systemic administration of SR-717 in a “closed” conformation | 2020 [28] |
| mouse | oral administration of MSA-2 in a “closed” conformation | 2020 [29] | ||
| mouse | engineered extracellular vesicle exogenously loaded with cyclic dinucleotide | 2021 [30] | ||
| human | intravenous infusion of TAK-676 | ongoing NCT04420884 | ||
| human | intravenous infusion of SB 11285 | ongoing NCT04096638 | ||
| human | intravenous infusion of SNX281 | ongoing NCT04609579 | ||
| human | intratumoral injection of CDK-002 | ongoing NCT04592484 | ||
| TLR9 | sensing tumor-derived DNA, DC maturation, and CTL priming | human | intratumoral injection of Vidutolimod | 2021 [24] |
| mouse | intratumoral injection CpG oligodeoxynucleotide (TLR9 ligand) and an antibody against OX40 | 2022 [70] | ||
| TLR3 | maturation and CTL priming | human | intraperitoneal injection of rintalomid (TLR3 agonist), celecoxib, and cisplatin | 2022 [60] |
| VEGF | local lymphangiogenesis, immune cell trafficking, and CTL activation | mouse | injection of “VEGFC vax” | 2021 [35] |
| XCL1+ Flt3L | DC recruitment and expansion | mouse | intratumoral injection of XCL1 and SFlt3L encoded in recombinant Semliki Forest virus-derived vectors | 2018 [50] |
| CCL4 | DC recruitment | mouse | intravenous administration of a fusion protein of CCL4 and the collagen-binding domain of von Willebrand factor | 2019 [51] |
| CXCL9/10/11 | CTL recruitment | mouse | CXCL9 and OX40L | 2020 [53] |
| mouse | intravenous injection of oncolytic vesicular stomatitis virus encodes CXCL9 | 2020 [55] | ||
| mouse | genetically engineered mesenchymal stem cells producing CXCL10 | 2018 [71] | ||
| humanized mouse | injection of CXCL10-producing SynNotch T cells | 2021 [72] | ||
| mouse | intravenous delivery of CXCL9/10/11 plasmids by nanoparticles | 2022 [73] | ||
| human | NG-641 is an oncolytic adenoviral vector which expresses a FAP-TAc antibody together with an immune enhancer module (CXCL9/CXCL10/IFNα). | ongoing NCT04053283 | ||
| IL12 | DC activation/IFNγ production of T and NK cells | mouse | intravenous injection of IL12 fused to a domain of the IL12 receptor | 2022 [61] |
| IL15 | DC recruitment/activation and downstream recruitment of CTLs/NK cells | mouse | intraperitoneal injection of heterodimeric IL-15 | 2020 [47] |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The Immune Contexture in Human Tumours: Impact on Clinical Outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine Expression in Melanoma Metastases Associated with CD8 + T-Cell Recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef]
- Liu, J.; Geng, X.; Hou, J.; Wu, G. New Insights into M1/M2 Macrophages: Key Modulators in Cancer Progression. Cancer Cell Int. 2021, 21, 389. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of CDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-Intrinsic β-Catenin Signalling Prevents Anti-Tumour Immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic Cells, Monocytes and Macrophages: A Unified Nomenclature Based on Ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of Immunogenic Antigens Does Not Explain the Presence or Absence of the T-Cell–Inflamed Tumor Microenvironment in Melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7759–E7768. [Google Scholar] [CrossRef]
- Flood, B.A.; Higgs, E.F.; Li, S.; Luke, J.J.; Gajewski, T.F. STING Pathway Agonism as a Cancer Therapeutic. Immunol. Rev. 2019, 290, 24–38. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like Receptor 4–Dependent Contribution of the Immune System to Anticancer Chemotherapy and Radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Kang, T.H.; Mao, C.-P.; Lee, S.Y.; Chen, A.; Lee, J.-H.; Kim, T.W.; Alvarez, R.D.; Roden, R.B.S.; Pardoll, D.; Hung, C.-F.; et al. Chemotherapy Acts as an Adjuvant to Convert the Tumor Microenvironment into a Highly Permissive State for Vaccination-Induced Antitumor Immunity. Cancer Res. 2013, 73, 2493–2504. [Google Scholar] [CrossRef]
- Kang, T.H.; Mao, C.-P.; Kim, Y.S.; Kim, T.W.; Yang, A.; Lam, B.; Tseng, S.-H.; Farmer, E.; Park, Y.-M.; Hung, C.-F. TLR9 Acts as a Sensor for Tumor-Released DNA to Modulate Anti-Tumor Immunity after Chemotherapy. J. Immunother. Cancer 2019, 7, 260. [Google Scholar] [CrossRef] [PubMed]
- Loef, E.J.; Sheppard, H.M.; Birch, N.P.; Dunbar, P.R. Live-Cell Microscopy Reveals That Human T Cells Primarily Respond Chemokinetically Within a CCL19 Gradient That Induces Chemotaxis in Dendritic Cells. Front. Immunol. 2021, 12, 863. [Google Scholar] [CrossRef] [PubMed]
- Martín-Fontecha, A.; Sebastiani, S.; Höpken, U.E.; Uguccioni, M.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Regulation of Dendritic Cell Migration to the Draining Lymph Node: Impact on T Lymphocyte Traffic and Priming. J. Exp. Med. 2003, 198, 615–621. [Google Scholar] [CrossRef]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Matson, V.; Flood, B.; Spranger, S.; Gajewski, T.F. Innate Immune Signaling and Regulation in Cancer Immunotherapy. Cell Res. 2017, 27, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Roberts, E.W.; Cai, E.; Mujal, A.M.; Marchuk, K.; Beppler, C.; Nam, D.; Serwas, N.K.; Binnewies, M.; Krummel, M.F. Visualizing Synaptic Transfer of Tumor Antigens among Dendritic Cells. Cancer Cell 2020, 37, 786–799.e5. [Google Scholar] [CrossRef]
- Duong, E.; Fessenden, T.B.; Lutz, E.; Dinter, T.; Yim, L.; Blatt, S.; Bhutkar, A.; Wittrup, K.D.; Spranger, S. Type I Interferon Activates MHC Class I-Dressed CD11b+ Conventional Dendritic Cells to Promote Protective Anti-Tumor CD8+ T Cell Immunity. Immunity 2022, 55, 308–323.e9. [Google Scholar] [CrossRef]
- Shulman, Z.; Cohen, S.J.; Roediger, B.; Kalchenko, V.; Jain, R.; Grabovsky, V.; Klein, E.; Shinder, V.; Stoler-Barak, L.; Feigelson, S.W.; et al. Transendothelial Migration of Lymphocytes Mediated by Intraendothelial Vesicle Stores Rather than by Extracellular Chemokine Depots. Nat. Immunol. 2012, 13, 67–76. [Google Scholar] [CrossRef]
- Franciszkiewicz, K.; Le Floc’h, A.; Boutet, M.; Vergnon, I.; Schmitt, A.; Mami-Chouaib, F. CD103 or LFA-1 Engagement at the Immune Synapse between Cytotoxic T Cells and Tumor Cells Promotes Maturation and Regulates T-Cell Effector Functions. Cancer Res. 2013, 73, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Ribas, A.; Medina, T.; Kirkwood, J.M.; Zakharia, Y.; Gonzalez, R.; Davar, D.; Chmielowski, B.; Campbell, K.M.; Bao, R.; Kelley, H.; et al. Overcoming PD-1 Blockade Resistance With CpG-A Toll-Like Receptor 9 Agonist Vidutolimod in Patients With Metastatic Melanoma. Cancer Discov. 2021, 11, 2998–3007. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.; Burdette, D.L.; Sharma, S.; Bhat, N.; Thompson, M.; Jiang, Z.; Rathinam, V.A.K.; Monks, B.; Jin, T.; Xiao, T.S.; et al. Mouse, but Not Human STING, Binds and Signals in Response to the Vascular Disrupting Agent 5,6-Dimethylxanthenone-4-Acetic Acid. J. Immunol. 2013, 190, 5216–5225. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Higgs, E.F. Immunotherapy with a Sting. Science 2020, 369, 921–922. [Google Scholar] [CrossRef]
- Chin, E.N.; Yu, C.; Vartabedian, V.F.; Jia, Y.; Kumar, M.; Gamo, A.M.; Vernier, W.; Ali, S.H.; Kissai, M.; Lazar, D.C.; et al. Antitumor Activity of a Systemic STING-Activating Non-Nucleotide CGAMP Mimetic. Science 2020, 369, 993–999. [Google Scholar] [CrossRef]
- Pan, B.-S.; Perera, S.A.; Piesvaux, J.A.; Presland, J.P.; Schroeder, G.K.; Cumming, J.N.; Trotter, B.W.; Altman, M.D.; Buevich, A.V.; Cash, B.; et al. An Orally Available Non-Nucleotide STING Agonist with Antitumor Activity. Science 2020, 369, eaba6098. [Google Scholar] [CrossRef]
- Jang, S.C.; Economides, K.D.; Moniz, R.J.; Sia, C.L.; Lewis, N.; McCoy, C.; Zi, T.; Zhang, K.; Harrison, R.A.; Lim, J.; et al. ExoSTING, an Extracellular Vesicle Loaded with STING Agonists, Promotes Tumor Immune Surveillance. Commun. Biol. 2021, 4, 1–17. [Google Scholar] [CrossRef]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING Activation of Tumor Endothelial Cells Initiates Spontaneous and Therapeutic Antitumor Immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lee, W.S.; Kong, S.J.; Kim, C.G.; Kim, J.H.; Chang, S.K.; Kim, S.; Kim, G.; Chon, H.J.; Kim, C. STING Activation Reprograms Tumor Vasculatures and Synergizes with VEGFR2 Blockade. J. Clin. Investig. 2019, 129, 4350–4364. [Google Scholar] [CrossRef] [PubMed]
- Spaapen, R.M.; Leung, M.Y.K.; Fuertes, M.B.; Kline, J.P.; Zhang, L.; Zheng, Y.; Fu, Y.-X.; Luo, X.; Cohen, K.S.; Gajewski, T.F. Therapeutic Activity of High-Dose Intratumoral IFN-β Requires Direct Effect on the Tumor Vasculature. J. Immunol. 2014, 193, 4254–4260. [Google Scholar] [CrossRef]
- Lund, A.W.; Wagner, M.; Fankhauser, M.; Steinskog, E.S.; Broggi, M.A.; Spranger, S.; Gajewski, T.F.; Alitalo, K.; Eikesdal, H.P.; Wiig, H.; et al. Lymphatic Vessels Regulate Immune Microenvironments in Human and Murine Melanoma. J. Clin. Investig. 2016, 126, 3389–3402. [Google Scholar] [CrossRef]
- Sasso, M.S.; Mitrousis, N.; Wang, Y.; Briquez, P.S.; Hauert, S.; Ishihara, J.; Hubbell, J.A.; Swartz, M.A. Lymphangiogenesis-Inducing Vaccines Elicit Potent and Long-Lasting T Cell Immunity against Melanomas. Sci. Adv. 2021, 7, eabe4362. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Woo, S.-R.; Burnett, B.; Fu, Y.-X.; Gajewski, T.F. Type I Interferon Response and Innate Immune Sensing of Cancer. Trends Immunol. 2013, 34, 67–73. [Google Scholar] [CrossRef]
- House, I.G.; Savas, P.; Lai, J.; Chen, A.X.Y.; Oliver, A.J.; Teo, Z.L.; Todd, K.L.; Henderson, M.A.; Giuffrida, L.; Petley, E.V.; et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin. Cancer Res. 2020, 26, 487–504. [Google Scholar] [CrossRef]
- Reschke, R.; Yu, J.; Flood, B.A.; Higgs, E.F.; Hatogai, K.; Gajewski, T.F. Immune Cell and Tumor Cell-Derived CXCL10 Is Indicative of Immunotherapy Response in Metastatic Melanoma. J. Immunother. Cancer 2021, 9, e003521. [Google Scholar] [CrossRef]
- Aliberti, J.; Reis e Sousa, C.; Schito, M.; Hieny, S.; Wells, T.; Huffnagle, G.B.; Sher, A. CCR5 Provides a Signal for Microbial Induced Production of IL-12 by CD8 Alpha+ Dendritic Cells. Nat. Immunol. 2000, 1, 83–87. [Google Scholar] [CrossRef]
- Dorner, B.G.; Dorner, M.B.; Zhou, X.; Opitz, C.; Mora, A.; Güttler, S.; Hutloff, A.; Mages, H.W.; Ranke, K.; Schaefer, M.; et al. Selective Expression of the Chemokine Receptor XCR1 on Cross-Presenting Dendritic Cells Determines Cooperation with CD8+ T Cells. Immunity 2009, 31, 823–833. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.-R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host Type I IFN Signals Are Required for Antitumor CD8+ T Cell Responses through CD8α+ Dendritic Cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; De Marco, C.; Cristiani, C.M. K Cells in the Tumor Microenvironment as New Potential Players Mediating Chemotherapy Effects in Metastatic Melanoma. N. Front. Oncol. 2021, 12, 754541. [Google Scholar] [CrossRef]
- McKay, K.; Moore, P.C.; Smoller, B.R.; Hiatt, K.M. Association between natural killer cells and regression in melanocytic lesions. Hum. Pathol. 2011, 42, 1960–1964. [Google Scholar] [CrossRef]
- Reschke, R.; Dumann, K.; Ziemer, M. Risk Stratification and Clinical Characteristics of Patients with Late Recurrence of Melanoma (>10 Years). J. Clin. Med. 2022, 11, 2026. [Google Scholar] [CrossRef]
- Mittal, D.; Vijayan, D.; Putz, E.M.; Aguilera, A.R.; Markey, K.A.; Straube, J.; Kazakoff, S.; Nutt, S.L.; Takeda, K.; Hill, G.R.; et al. Interleukin-12 from CD103+ Batf3-Dependent Dendritic Cells Required for NK-Cell Suppression of Metastasis. Cancer Immunol. Res. 2017, 5, 1098–1108. [Google Scholar] [CrossRef]
- Mattei, F.; Schiavoni, G.; Belardelli, F.; Tough, D.F. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J. Immunol. 2001, 167, 1179–1187. [Google Scholar] [CrossRef]
- Bergamaschi, C.; Pandit, H.; Nagy, B.A.; Stellas, D.; Jensen, S.M.; Bear, J.; Cam, M.; Valentin, A.; Fox, B.A.; Felber, B.K.; et al. Heterodimeric IL-15 delays tumor growth and promotes intratumoral CTL and dendritic cell accumulation by a cytokine network involving XCL1, IFN-γ, CXCL9 and CXCL10. J. Immunother. Cancer 2020, 8, e000599. [Google Scholar] [CrossRef]
- Reschke, R.; Gajewski, T.F. CXCL9 and CXCL10 bring the heat to tumors. Sci. Immunol. 2022, 7, eabq6509. [Google Scholar] [CrossRef]
- Hoch, T.; Schulz, D.; Eling, N.; Gómez, J.M.; Levesque, M.P.; Bodenmiller, B. Multiplexed imaging mass cytometry of the chemokine milieus in melanoma characterizes features of the response to immunotherapy. Sci. Immunol. 2022, 7, eabk1692. [Google Scholar] [CrossRef]
- Sánchez-Paulete, A.R.; Teijeira, Á.; Quetglas, J.I.; Rodríguez-Ruiz, M.E.; Sánchez-Arráez, Á.; Labiano, S.; Etxeberria, I.; Azpilikueta, A.; Bolaños, E.; Ballesteros-Briones, M.C.; et al. Intratumoral Immunotherapy with XCL1 and SFlt3L Encoded in Recombinant Semliki Forest Virus–Derived Vectors Fosters Dendritic Cell–Mediated T-Cell Cross-Priming. Cancer Res. 2018, 78, 6643–6654. [Google Scholar] [CrossRef]
- Williford, J.-M.; Ishihara, J.; Ishihara, A.; Mansurov, A.; Hosseinchi, P.; Marchell, T.M.; Potin, L.; Swartz, M.A.; Hubbell, J.A. Recruitment of CD103+ Dendritic Cells via Tumor-Targeted Chemokine Delivery Enhances Efficacy of Checkpoint Inhibitor Immunotherapy. Sci. Adv. 2019, 5, eaay1357. [Google Scholar] [CrossRef] [PubMed]
- Antonicelli, F.; Lorin, J.; Kurdykowski, S.; Gangloff, S.C.; Le Naour, R.; Sallenave, J.M.; Hornebeck, W.; Grange, F.; Bernard, P. CXCL10 Reduces Melanoma Proliferation and Invasiveness in Vitro and in Vivo: CXCL10 and Melanoma Progression. Br. J. Dermatol. 2011, 164, 720–728. [Google Scholar] [CrossRef]
- Yin, P.; Gui, L.; Wang, C.; Yan, J.; Liu, M.; Ji, L.; Wang, Y.; Ma, B.; Gao, W.-Q. Targeted Delivery of CXCL9 and OX40L by Mesenchymal Stem Cells Elicits Potent Antitumor Immunity. Mol. Ther. 2020, 28, 2553–2563. [Google Scholar] [CrossRef] [PubMed]
- Cervera-Carrascon, V.; Quixabeira, D.C.A.; Santos, J.M.; Havunen, R.; Zafar, S.; Hemminki, O.; Heiniö, C.; Munaro, E.; Siurala, M.; Sorsa, S.; et al. Tumor Microenvironment Remodeling by an Engineered Oncolytic Adenovirus Results in Improved Outcome from PD-L1 Inhibition. OncoImmunology 2020, 9, 1761229. [Google Scholar] [CrossRef]
- Eckert, E.C.; Nace, R.A.; Tonne, J.M.; Evgin, L.; Vile, R.G.; Russell, S.J. Generation of a Tumor-Specific Chemokine Gradient Using Oncolytic Vesicular Stomatitis Virus Encoding CXCL9. Mol. Ther. Oncolytics 2019, 16, 63–74. [Google Scholar] [CrossRef]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational Combination of Oncolytic Vaccinia Virus and PD-L1 Blockade Works Synergistically to Enhance Therapeutic Efficacy. Nat. Commun. 2017, 8, 14754. [Google Scholar] [CrossRef]
- Mowat, C.; Mosley, S.R.; Namdar, A.; Schiller, D.; Baker, K. Anti-Tumor Immunity in Mismatch Repair-Deficient Colorectal Cancers Requires Type I IFN–Driven CCL5 and CXCL10. J. Exp. Med. 2021, 218, e20210108. [Google Scholar] [CrossRef]
- Kobayashi, H.; Nobeyama, Y.; Nakagawa, H. Tumor-Suppressive Effects of Natural-Type Interferon-β through CXCL10 in Melanoma. Biochem. Biophys. Res. Commun. 2015, 464, 416–421. [Google Scholar] [CrossRef]
- Peng, W.; Liu, C.; Xu, C.; Lou, Y.; Chen, J.; Yang, Y.; Yagita, H.; Overwijk, W.W.; Lizée, G.; Radvanyi, L.; et al. PD-1 blockade enhances T cell migration to tumors by elevating IFN-γ inducible chemokines. Cancer Res. 2012, 72, 5209–5218. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.; Mahdi, H.; Fang, Y.; Strange, M.; Uygun, I.; Rana, M.; Zhang, L.; Mora, A.S.; Pusateri, A.; Elishaev, E.; et al. Phase I trial combining chemokine-targeting with loco-regional chemoimmunotherapy for recurrent, platinum-sensitive ovarian cancer shows induction of CXCR3 ligands and markers of type 1 immunity. Clin. Cancer Res. 2022, 28, 2038–2049. [Google Scholar]
- Mansurov, A.; Hosseinchi, P.; Chang, K.; Lauterbach, A.L.; Gray, L.T.; Alpar, A.T.; Budina, E.; Slezak, A.J.; Kang, S.; Cao, S.; et al. Masking the immunotoxicity of interleukin-12 by fusing it with a domain of its receptor via a tumour-protease-cleavable linker. Nat. Biomed. Eng. 2022, 6, 819–829. [Google Scholar] [CrossRef]
- Antonelli, A.; Ferrari, S.M.; Corrado, A.; Ferrannini, E.; Fallahi, P. CXCR3, CXCL10 and Type 1 Diabetes. Cytokine Growth Factor Rev. 2014, 25, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Boniface, K.; Jacquemin, C.; Darrigade, A.-S.; Dessarthe, B.; Martins, C.; Boukhedouni, N.; Vernisse, C.; Grasseau, A.; Thiolat, D.; Rambert, J.; et al. Vitiligo Skin Is Imprinted with Resident Memory CD8 T Cells Expressing CXCR3. J. Investig. Dermatol. 2018, 138, 355–364. [Google Scholar] [CrossRef]
- Han, J.H.; Suh, C.-H.; Jung, J.-Y.; Ahn, M.-H.; Han, M.H.; Kwon, J.E.; Yim, H.; Kim, H.-A. Elevated Circulating Levels of the Interferon-γ-Induced Chemokines Are Associated with Disease Activity and Cutaneous Manifestations in Adult-Onset Still’s Disease. Sci. Rep. 2017, 7, 46652. [Google Scholar] [CrossRef]
- Ito, T. Recent Advances in the Pathogenesis of Autoimmune Hair Loss Disease Alopecia Areata. Clin. Dev. Immunol. 2013, 2013, 348546. [Google Scholar] [CrossRef] [PubMed]
- Loos, T.; Dekeyzer, L.; Struyf, S.; Schutyser, E.; Gijsbers, K.; Gouwy, M.; Fraeyman, A.; Put, W.; Ronsse, I.; Grillet, B.; et al. TLR Ligands and Cytokines Induce CXCR3 Ligands in Endothelial Cells: Enhanced CXCL9 in Autoimmune Arthritis. Lab. Investig. 2006, 86, 902–916. [Google Scholar] [CrossRef] [PubMed]
- Luoma, A.M.; Suo, S.; Williams, H.L.; Sharova, T.; Sullivan, K.; Manos, M.; Bowling, P.; Hodi, F.S.; Rahma, O.; Sullivan, R.J.; et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 2020, 182, 655–671.e22. [Google Scholar] [CrossRef]
- Reschke, R.; Gussek, P.; Boldt, A.; Sack, U.; Köhl, U.; Lordick, F.; Gora, T.; Kreuz, M.; Reiche, K.; Simon, J.C.; et al. Distinct Immune Signatures Indicative of Treatment Response and Immune-Related Adverse Events in Melanoma Patients under Immune Checkpoint Inhibitor Therapy. Int. J. Mol. Sci. 2021, 22, 8017. [Google Scholar] [CrossRef]
- Amatschek, S.; Lucas, R.; Eger, A.; Pflueger, M.; Hundsberger, H.; Knoll, C.; Grosse-Kracht, S.; Schuett, W.; Koszik, F.; Maurer, D.; et al. CXCL9 Induces Chemotaxis, Chemorepulsion and Endothelial Barrier Disruption through CXCR3-Mediated Activation of Melanoma Cells. Br. J. Cancer 2011, 104, 469–479. [Google Scholar] [CrossRef]
- Hong, W.X.; Sagiv-Barfi, I.; Czerwinski, D.K.; Sallets, A.; Levy, R. Neoadjuvant Intratumoral Immunotherapy with TLR9 Activation and Anti-OX40 Antibody Eradicates Metastatic Cancer. Cancer Res. 2022, 82, 1396–1408. [Google Scholar] [CrossRef]
- Mirzaei, H.; Salehi, H.; Oskuee, R.K.; Mohammadpour, A.; Mirzaei, H.R.; Sharifi, M.R.; Salarinia, R.; Darani, H.Y.; Mokhtari, M.; Masoudifar, A.; et al. The Therapeutic Potential of Human Adipose-Derived Mesenchymal Stem Cells Producing CXCL10 in a Mouse Melanoma Lung Metastasis Model. Cancer Lett. 2018, 419, 30–39. [Google Scholar] [CrossRef]
- Xia, M.; Chen, J.; Meng, G.; Shen, H.; Dong, J. CXCL10 Encoding SynNotch T Cells Enhance Anti-Tumor Immune Responses without Systemic Side Effect. Biochem. Biophys. Res. Commun. 2021, 534, 765–772. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, Y.; Zhi, Y.; Wang, H.; Yang, M.; Niu, J.; Zhao, L.; Wang, P. Delivery of CXCL9/10/11 Plasmid DNAs Promotes the Tumor-Infiltration of T Cells and Synergizes with PD1 Antibody for Treating Lung Cancer. Cancer Nanotechnol. 2022, 13, 10. [Google Scholar] [CrossRef]
- Reschke, R.; Jäger, I.; Mehnert-Theuerkauf, A.; Ziemer, M. Therapy understanding and health related quality of life in stage III/IV melanoma patients treated with novel adjuvant therapies. J. Dtsch. Dermatol. Ges. 2021, 19, 215–221. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reschke, R.; Olson, D.J. Leveraging STING, Batf3 Dendritic Cells, CXCR3 Ligands, and Other Components Related to Innate Immunity to Induce a “Hot” Tumor Microenvironment That Is Responsive to Immunotherapy. Cancers 2022, 14, 2458. https://doi.org/10.3390/cancers14102458
Reschke R, Olson DJ. Leveraging STING, Batf3 Dendritic Cells, CXCR3 Ligands, and Other Components Related to Innate Immunity to Induce a “Hot” Tumor Microenvironment That Is Responsive to Immunotherapy. Cancers. 2022; 14(10):2458. https://doi.org/10.3390/cancers14102458
Chicago/Turabian StyleReschke, Robin, and Daniel J. Olson. 2022. "Leveraging STING, Batf3 Dendritic Cells, CXCR3 Ligands, and Other Components Related to Innate Immunity to Induce a “Hot” Tumor Microenvironment That Is Responsive to Immunotherapy" Cancers 14, no. 10: 2458. https://doi.org/10.3390/cancers14102458
APA StyleReschke, R., & Olson, D. J. (2022). Leveraging STING, Batf3 Dendritic Cells, CXCR3 Ligands, and Other Components Related to Innate Immunity to Induce a “Hot” Tumor Microenvironment That Is Responsive to Immunotherapy. Cancers, 14(10), 2458. https://doi.org/10.3390/cancers14102458