
Diphenyleneiodonium Triggers Cell Death of Acute Myeloid Leukemia Cells by Blocking the Mitochondrial Respiratory Chain, and Synergizes with Cytarabine

 , ,
, ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
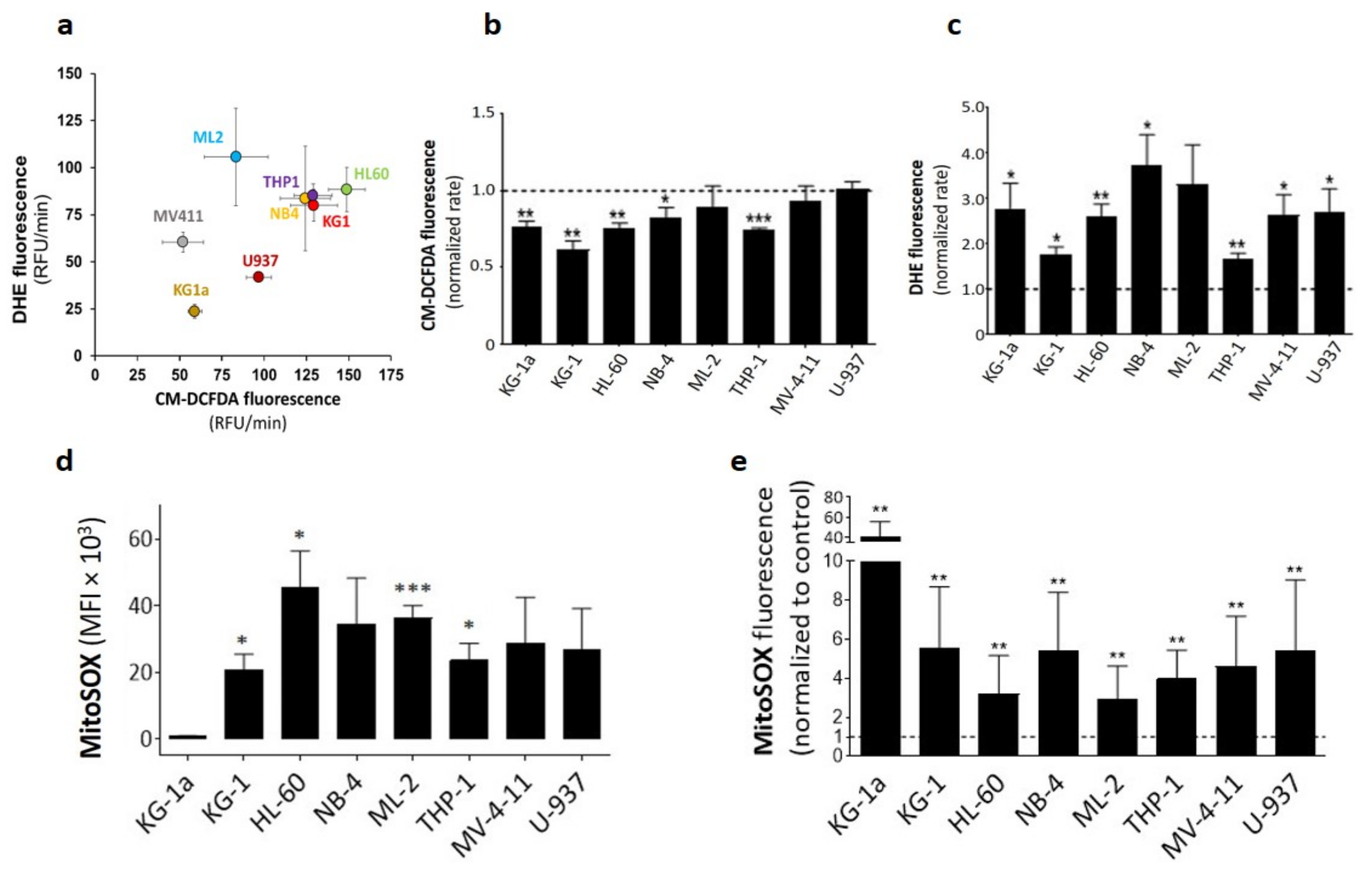
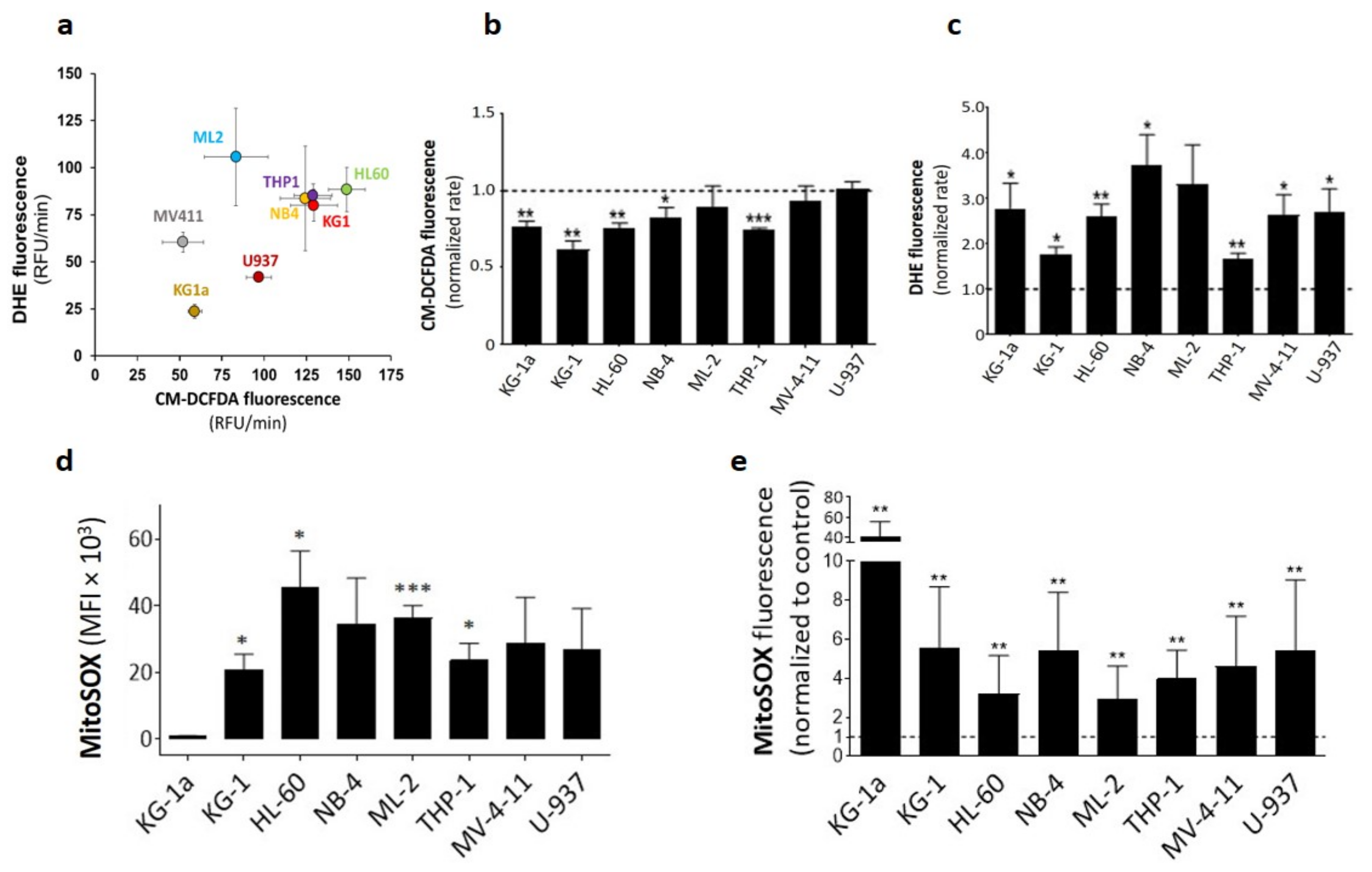
2.1. DPI Reduces Cytoplasmic ROS while Inducing Superoxide Production
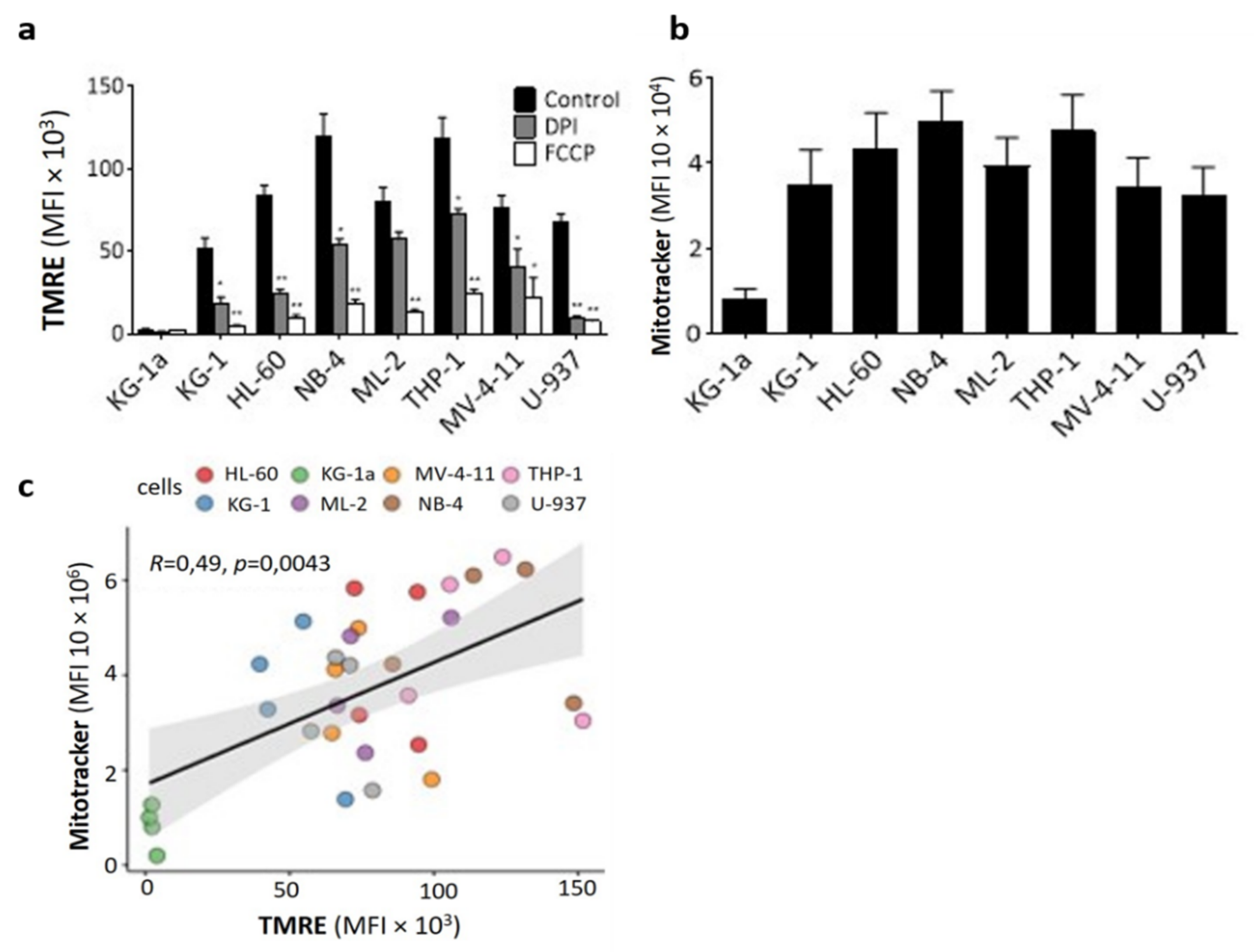
2.2. DPI Disrupts the Mitochondrial Membrane Potential
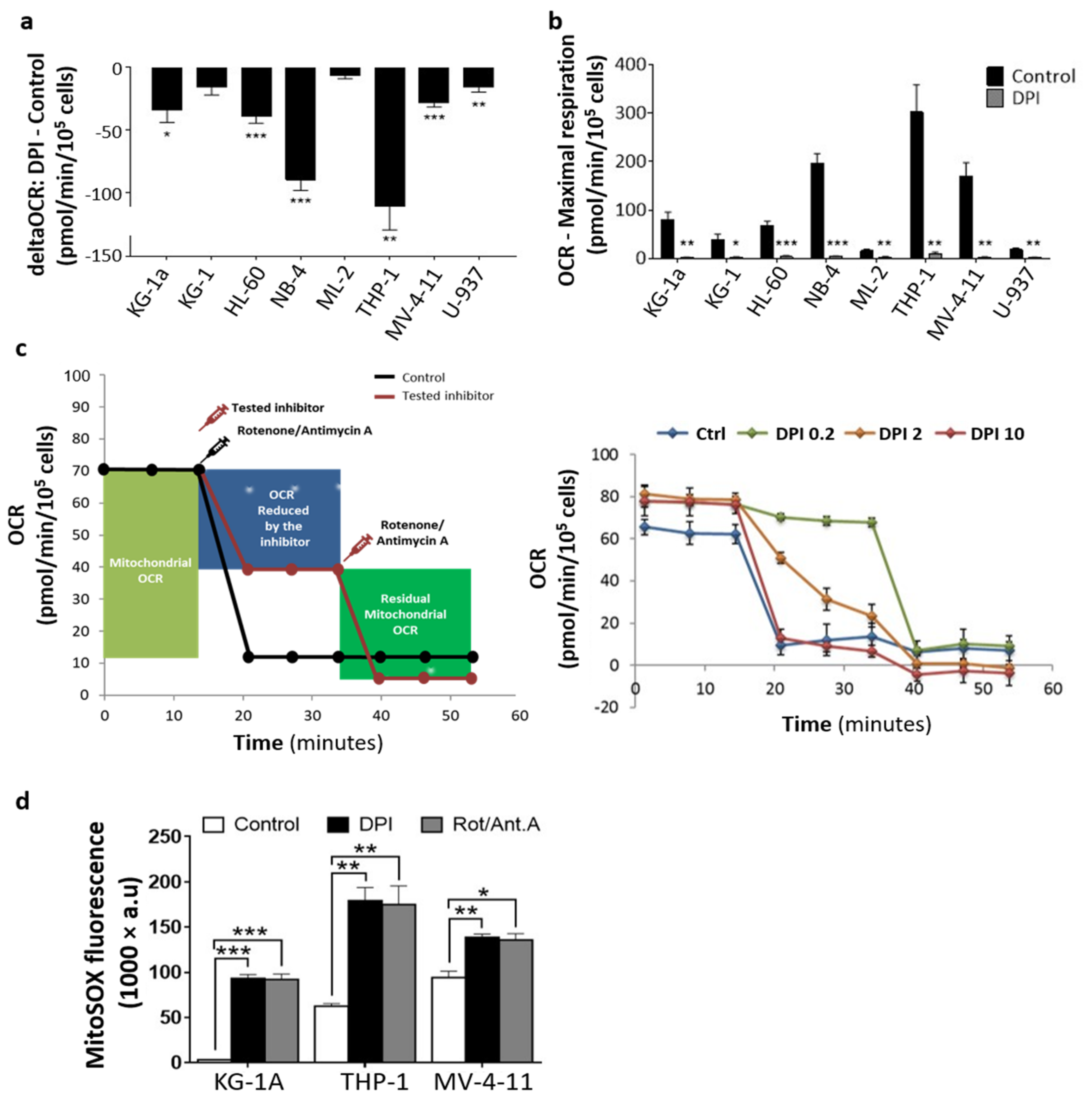
2.3. DPI Disrupts the MRC and Alters the Energetic Metabolism
2.4. DPI Reduces Cell Proliferation and Triggers Apoptosis
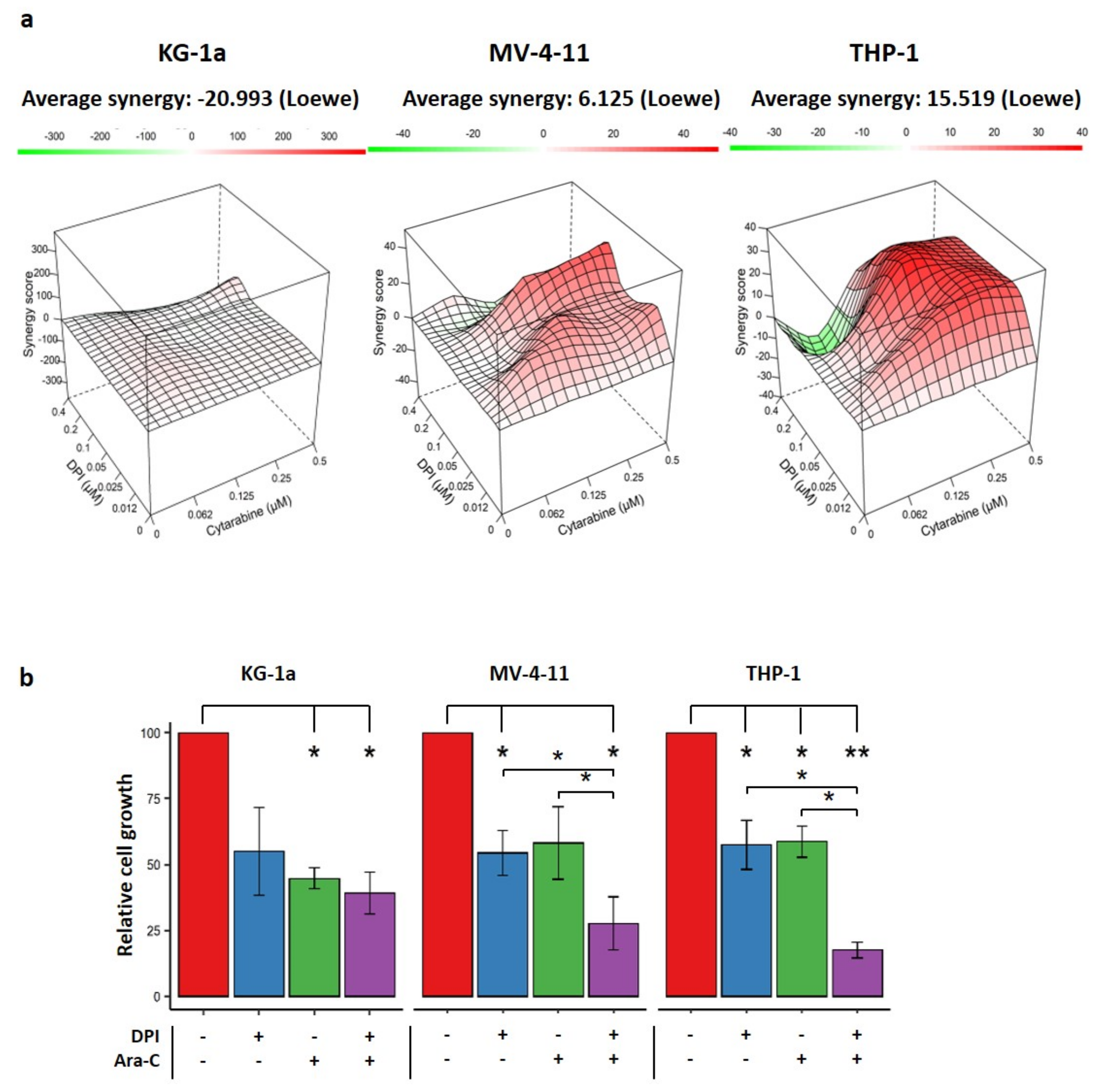
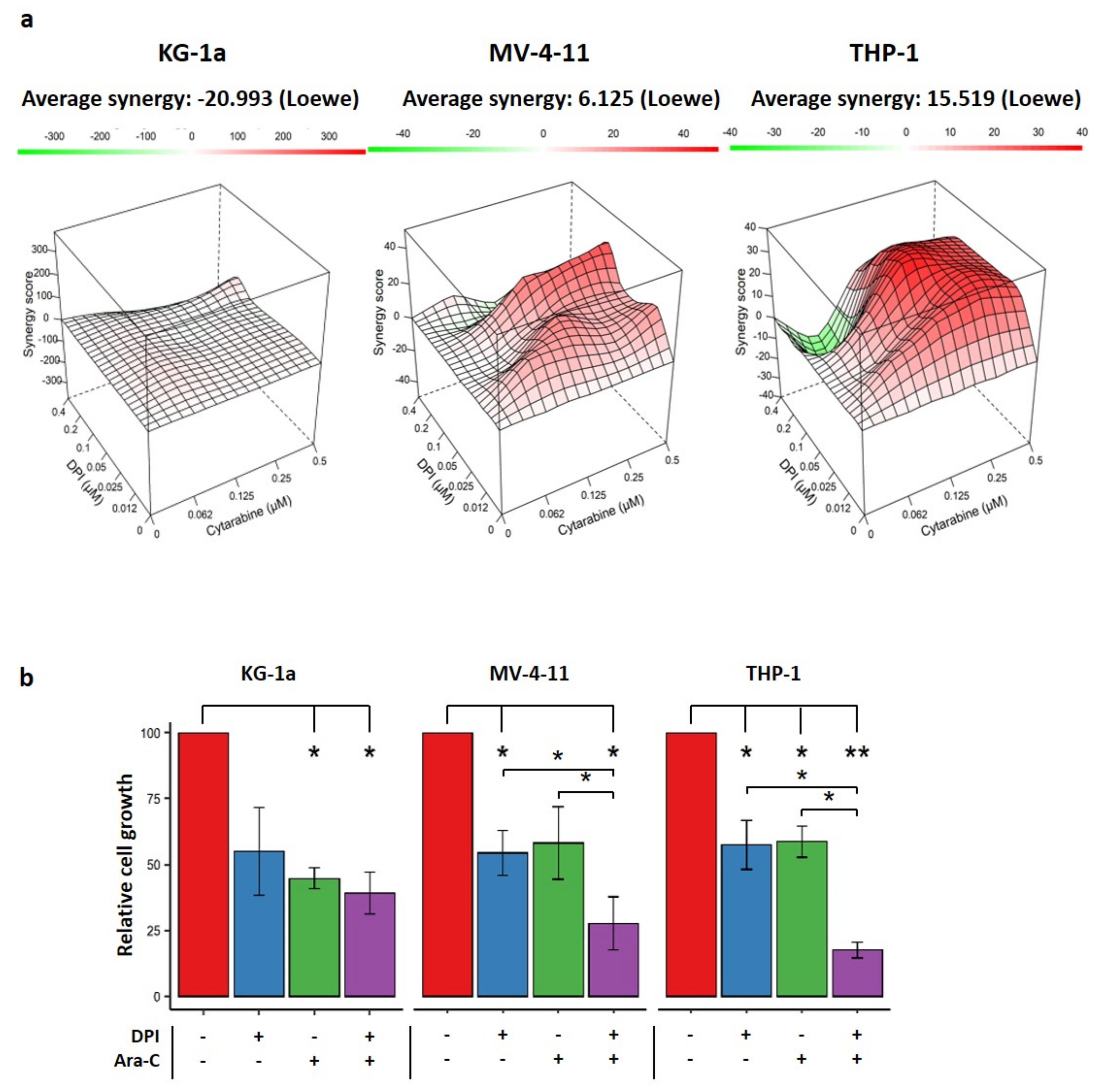
2.5. Effects of Combination Therapy of DPI and Cytarabine on AML Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. ROS Measurement
4.2.1. Fluorometric Assay (CM-DCFDA and DHE)
4.2.2. Flow Cytometry Assay (MitoSOX)
4.3. Mitochondrial Function Measurement
4.3.1. Mitochondrial Mass
4.3.2. Mitochondrial Membrane Potential
4.4. Mitochondrial Respiration
4.5. Apoptosis Assay
4.6. Drug Combination Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, T.K.; Wald, D.; Chen, Y.C.; Vermaat, J.M.; Xiong, Y.; Tse, W. Molecular prognostic markers for adult acute myeloid leukemia with normal cytogenetics. J. Hematol. Oncol. 2009, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, A.L.; Aslostovar, L.; Reid, J.; Ye, W.; Tanasijevic, B.; Porras, D.P.; Shapovalova, Z.; Almakadi, M.; Foley, R.; Leber, B.; et al. Identification of Chemotherapy-Induced Leukemic-Regenerating Cells Reveals a Transient Vulnerability of Human AML Recurrence. Cancer Cell 2018, 34, 483–498.e5. [Google Scholar] [CrossRef] [Green Version]
- Herault, O.; Hope, K.J.; Deneault, E.; Mayotte, N.; Chagraoui, J.; Wilhelm, B.T.; Cellot, S.; Sauvageau, M.; Andrade-Navarro, M.A.; Hébert, J.; et al. A role for GPx3 in activity of normal and leukemia stem cells. J. Exp. Med. 2012, 209, 895–901. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Trombetti, S.; Cesaro, E.; Catapano, R.; Sessa, R.; Lo Bianco, A.; Izzo, P.; Grosso, M. Oxidative Stress and ROS-Mediated Signaling in Leukemia: Novel Promising Perspectives to Eradicate Chemoresistant Cells in Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 2470. [Google Scholar] [CrossRef]
- Dakik, H.; El Dor, M.; Leclerc, J.; Kouzi, F.; Nehme, A.; Deynoux, M.; Debeissat, C.; Khamis, G.; Ducrocq, E.; Ibrik, A.; et al. Characterization of NADPH Oxidase Expression and Activity in Acute Myeloid Leukemia Cell Lines: A Correlation with the Differentiation Status. Antioxidants 2021, 10, 498. [Google Scholar] [CrossRef]
- Adane, B.; Ye, H.B.; Khan, N.; Pei, S.; Minhajuddin, M.; Stevens, B.M.; Jones, C.L.; D’Alessandro, A.; Reisz, J.A.; Zaberezhnyy, V.; et al. The Hematopoietic Oxidase NOX2 Regulates Self-Renewal of Leukemic Stem Cells. Cell Rep. 2019, 27, 238–254.e6. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes-Pagano, E.; Meijles, D.N.; Pagano, P.J. The Quest for Selective Nox Inhibitors and Therapeutics: Challenges, Triumphs and Pitfalls. Antioxid. Redox Signal. 2014, 20, 2741–2754. [Google Scholar] [CrossRef] [Green Version]
- Stuehr, D.J.; Fasehun, O.A.; Kwon, N.S.; Gross, S.S.; Gonzalez, J.A.; Levi, R.; Nathan, C.F. Inhibition of macrophage and endothelial cell nitric oxide synthase by diphenyleneiodonium and its analogs. FASEB J. 1991, 5, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.B.; Trush, M.A. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem. Biophys. Res. Commun. 1998, 253, 295–299. [Google Scholar] [CrossRef]
- Lambert, A.J.; Buckingham, J.A.; Boysen, H.M.; Brand, M.D. Diphenyleneiodonium acutely inhibits reactive oxygen species production by mitochondrial complex I during reverse, but not forward electron transport. Biochim. Biophys. Acta BBA—Bioenerg. 2008, 1777, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Tew, D.G. Inhibition of cytochrome P450 reductase by the diphenyliodonium cation. Kinetic analysis and covalent modifications. Biochemistry 1993, 32, 10209–10215. [Google Scholar] [CrossRef]
- Prabhakar, N.R. Oxygen sensing by the carotid body chemoreceptors. J. Appl. Physiol. 2000, 88, 2287–2295. [Google Scholar] [CrossRef] [Green Version]
- Doussiere, J.; Gaillard, J.; Vignais, P.V. The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry 1999, 38, 3694–3703. [Google Scholar] [CrossRef]
- Ozsvari, B.; Bonuccelli, G.; Sanchez-Alvarez, R.; Foster, R.; Sotgia, F.; Lisanti, M.P. Targeting flavin-containing enzymes eliminates cancer stem cells (CSCs), by inhibiting mitochondrial respiration: Vitamin B2 (Riboflavin) in cancer therapy. Aging 2017, 9, 2610–2628. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R.; Sultan, C. Proposals for classification of acute leukemias. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, B.; Gutierrez-Herrero, S.; Lopez-Ruano, G.; Prieto-Bermejo, R.; Romo-Gonzalez, M.; Llanillo, M.; Pandiella, A.; Guerrero, C.; Miguel, J.F.S.; Sanchez-Guijo, F.; et al. NADPH oxidases as therapeutic targets in chronic myelogenous leukemia. Clin. Cancer Res. 2014, 20, 4014–4025. [Google Scholar] [CrossRef] [Green Version]
- Odonnell, V.B.; Tew, D.G.; Jones, O.T.G.; England, P.J. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem. J. 1993, 290, 41–49. [Google Scholar] [CrossRef]
- Reddy, M.M.; Fernandes, M.S.; Salgia, R.; Levine, R.L.; Griffin, J.D.; Sattler, M. NADPH oxidases regulate cell growth and migration in myeloid cells transformed by oncogenic tyrosine kinases. Leukemia 2011, 25, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Brar, S.S.; Corbin, Z.; Kennedy, T.P.; Hemendinger, R.; Thornton, L.; Bommarius, B.; Arnold, R.S.; Whorton, A.R.; Sturrock, A.B.; Huecksteadt, T.P.; et al. NOX5 NAD(P)H oxidase regulates growth and apoptosis in DU 145 prostate cancer cells. Am. J. Physiol.—Cell Physiol. 2003, 285, C353–C369. [Google Scholar]
- Kumar, B.; Koul, S.; Khandrika, L.; Meacham, R.B.; Koul, H.K. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008, 68, 1777–1785. [Google Scholar] [CrossRef] [Green Version]
- El Dor, M.; Dakik, H.; Polomski, M.; Haudebourg, E.; Brachet, M.; Gouilleux, F.; Prie, G.; Zibara, K.; Mazurier, F. VAS3947 Induces UPR-Mediated Apoptosis through Cysteine Thiol Alkylation in AML Cell Lines. Int. J. Mol. Sci. 2020, 21, 5470. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Jayavelu, A.K.; Moloney, J.N.; Böhmer, F.-D.; Cotter, T.G. NOX-driven ROS formation in cell transformation of FLT3-ITD positive AML. Exp. Hematol. 2016, 44, 1113–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondet, J.; Lo Presti, C.; Garrel, C.; Skaare, K.; Mariette, C.; Carras, S.; Park, S.; Carre, M.; Bulabois, C.E.; Molina, L.; et al. Adult patients with de novo acute myeloid leukemia show a functional deregulation of redox balance at diagnosis which is correlated with molecular subtypes and overall survival. Haematologica 2019, 104, e393–e397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, A.J.; Hopkins, G.L.; Rastogi, N.; Hodges, M.; Doyle, M.; Davies, S.; Hole, P.S.; Omidvar, N.; Darley, R.L.; Tonks, A. Reactive Oxygen Species Drive Proliferation in Acute Myeloid Leukemia via the Glycolytic Regulator PFKFB3. Cancer Res. 2020, 80, 937–949. [Google Scholar] [CrossRef] [Green Version]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]
- Kouzi, F.; Zibara, K.; Bourgeais, J.; Picou, F.; Gallay, N.; Brossaud, J.; Dakik, H.; Roux, B.; Hamard, S.; Le Nail, L.R.; et al. Disruption of gap junctions attenuates acute myeloid leukemia chemoresistance induced by bone marrow mesenchymal stromal cells. Oncogene 2020, 39, 1198–1212. [Google Scholar] [CrossRef]
- He, L.; Kulesskiy, E.; Saarela, J.; Turunen, L.; Wennerberg, K.; Aittokallio, T.; Tang, J. Methods for High-throughput Drug Combination Screening and Synergy Scoring. Methods Mol. Biol. 2018, 1711, 351–398. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell line | Mitochondrial OCR IC50 | ||||||
|---|---|---|---|---|---|---|---|
| KG-1a | THP-1 | MV-4-11 | |||||
| Inhibitors | Target | IC50 (µM) | 95% CI (µM) | IC50 (µM) | 95% CI (µM) | IC50 (µM) | 95% CI (µM) |
| Rotenone | Complex I | 0.55 | 0.40 to 0.76 | 1.22 | 0.83 to 1.80 | 0.64 | 0.51 to 0.82 |
| Antimycin A | Complex III | 0.26 | 0.18 to 0.39 | 0.53 | 0.40 to 0.69 | 0.32 | 0.24 to 0.42 |
| DPI | Flavoproteins | 0.2 | 0.13 to 0.30 | 1.29 | 0.91 to 1.82 | 0.78 | 0.55 to 1.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dakik, H.; El Dor, M.; Bourgeais, J.; Kouzi, F.; Herault, O.; Gouilleux, F.; Zibara, K.; Mazurier, F. Diphenyleneiodonium Triggers Cell Death of Acute Myeloid Leukemia Cells by Blocking the Mitochondrial Respiratory Chain, and Synergizes with Cytarabine. Cancers 2022, 14, 2485. https://doi.org/10.3390/cancers14102485
Dakik H, El Dor M, Bourgeais J, Kouzi F, Herault O, Gouilleux F, Zibara K, Mazurier F. Diphenyleneiodonium Triggers Cell Death of Acute Myeloid Leukemia Cells by Blocking the Mitochondrial Respiratory Chain, and Synergizes with Cytarabine. Cancers. 2022; 14(10):2485. https://doi.org/10.3390/cancers14102485
Chicago/Turabian StyleDakik, Hassan, Maya El Dor, Jérôme Bourgeais, Farah Kouzi, Olivier Herault, Fabrice Gouilleux, Kazem Zibara, and Frédéric Mazurier. 2022. "Diphenyleneiodonium Triggers Cell Death of Acute Myeloid Leukemia Cells by Blocking the Mitochondrial Respiratory Chain, and Synergizes with Cytarabine" Cancers 14, no. 10: 2485. https://doi.org/10.3390/cancers14102485
APA StyleDakik, H., El Dor, M., Bourgeais, J., Kouzi, F., Herault, O., Gouilleux, F., Zibara, K., & Mazurier, F. (2022). Diphenyleneiodonium Triggers Cell Death of Acute Myeloid Leukemia Cells by Blocking the Mitochondrial Respiratory Chain, and Synergizes with Cytarabine. Cancers, 14(10), 2485. https://doi.org/10.3390/cancers14102485