
RON (MST1R) and HGFL (MST1) Co-Overexpression Supports Breast Tumorigenesis through Autocrine and Paracrine Cellular Crosstalk

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Histology and Immunohistochemistry (IHC)
2.3. Cell Cultures
2.4. Mammosphere Formation Assays
2.5. Migration Assays
2.6. Immunoblot Analyses and Cytokine Array
2.7. Bioinformatics
2.8. Statistics
3. Results
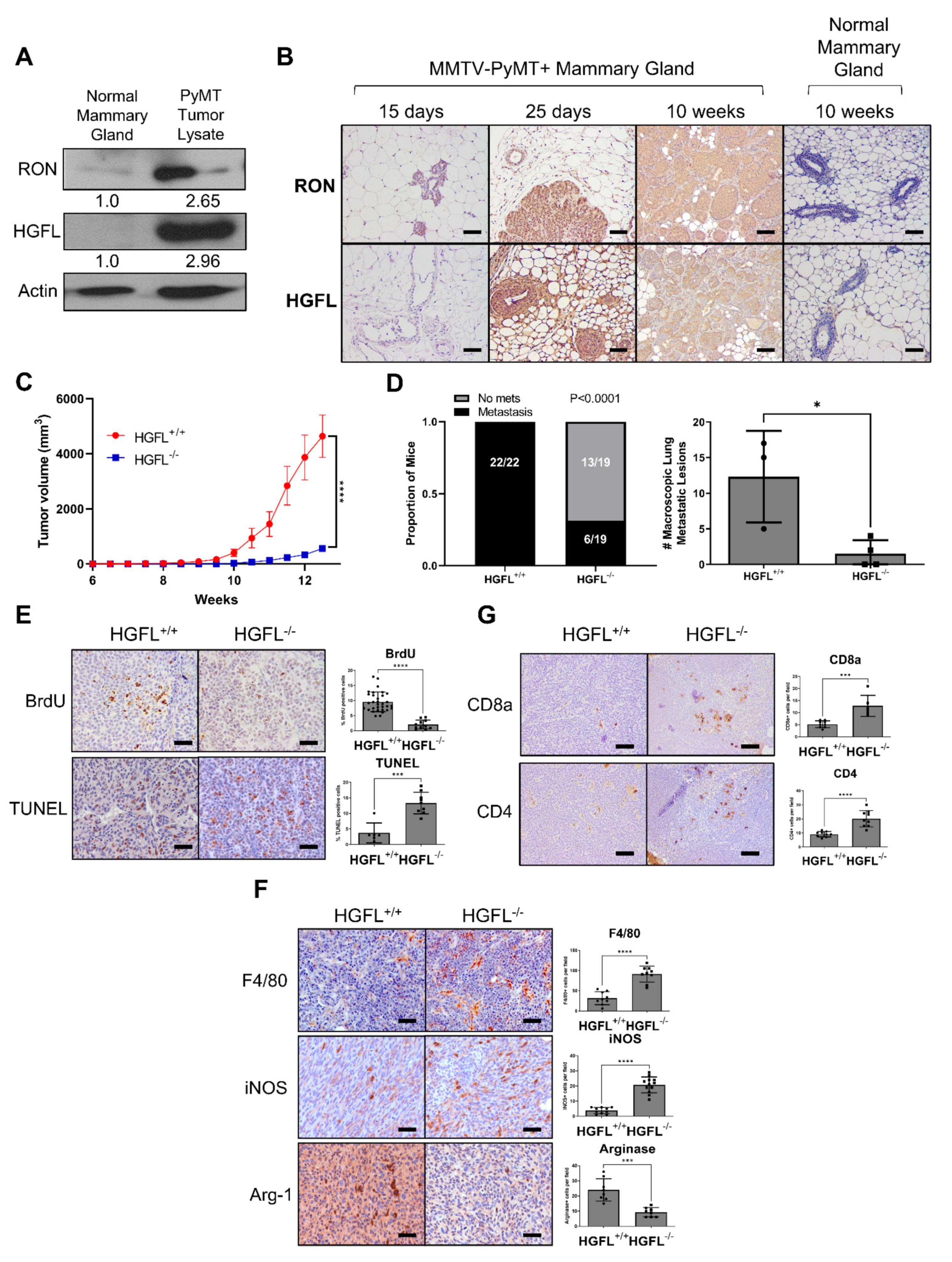
3.1. RON and HGFL Overexpression Co-Occurs in PyMT Tumors and Global HGFL Deletion Impairs Tumorigenesis, Metastatic Progression, and Improves T Cell Recruitment
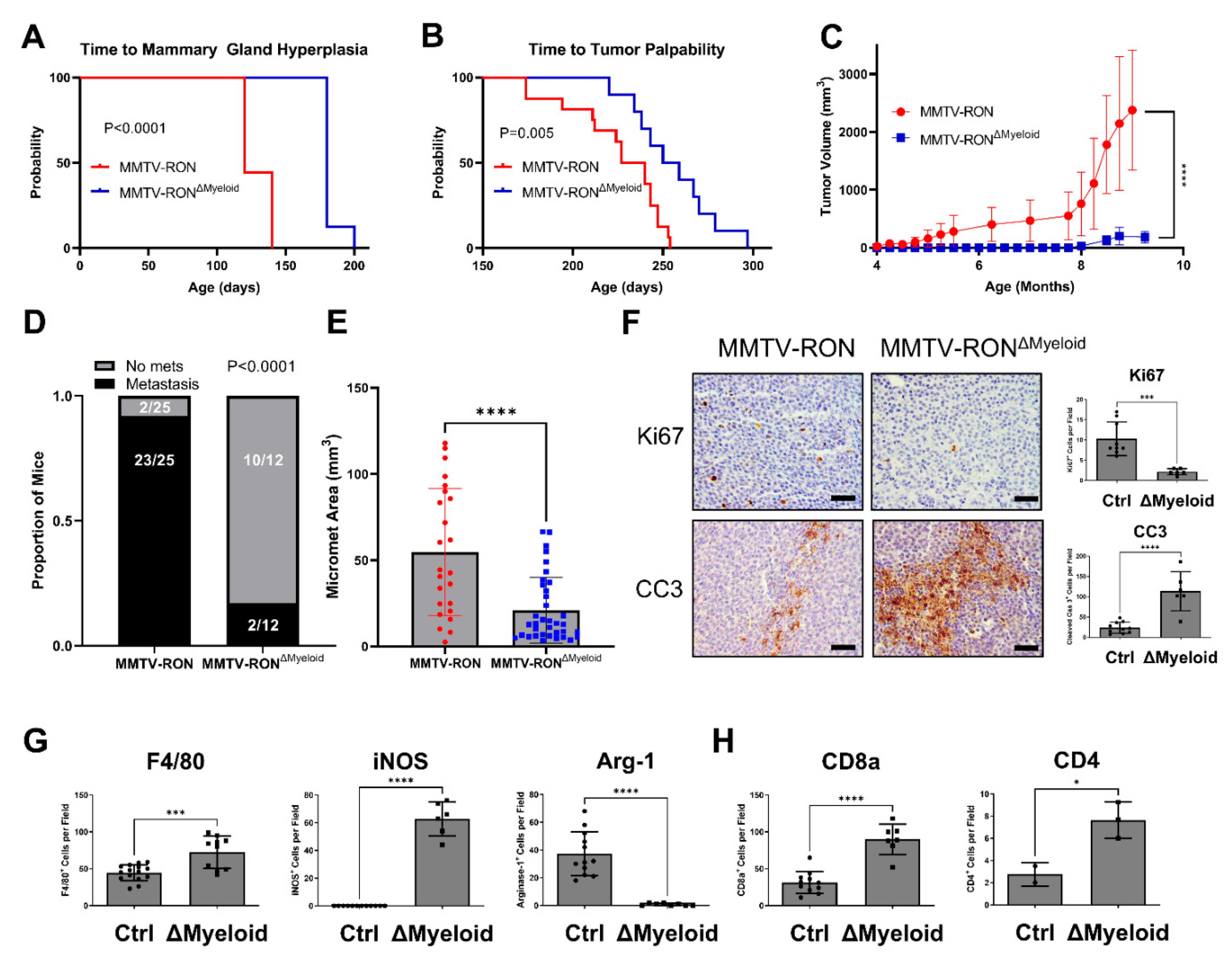
3.2. Conditional RON Loss in the Myeloid Compartment Phenocopies Global HGFL Loss in the RON-Driven Mammary Tumorigenesis Model, MMTV-RON
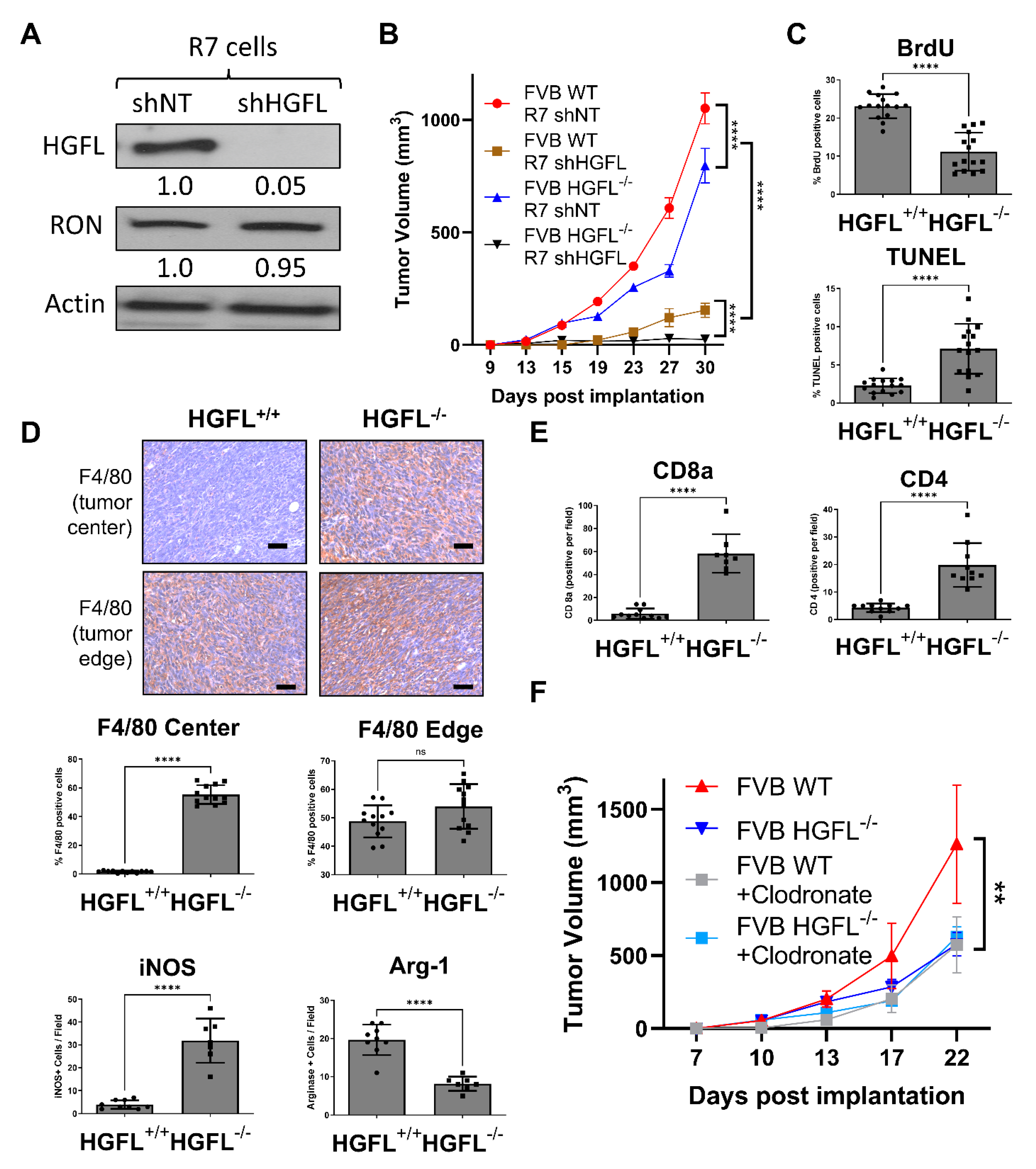
3.3. Tumor Cell-Secreted and Physiologic Sources of HGFL Each Support Mammary Tumor Growth
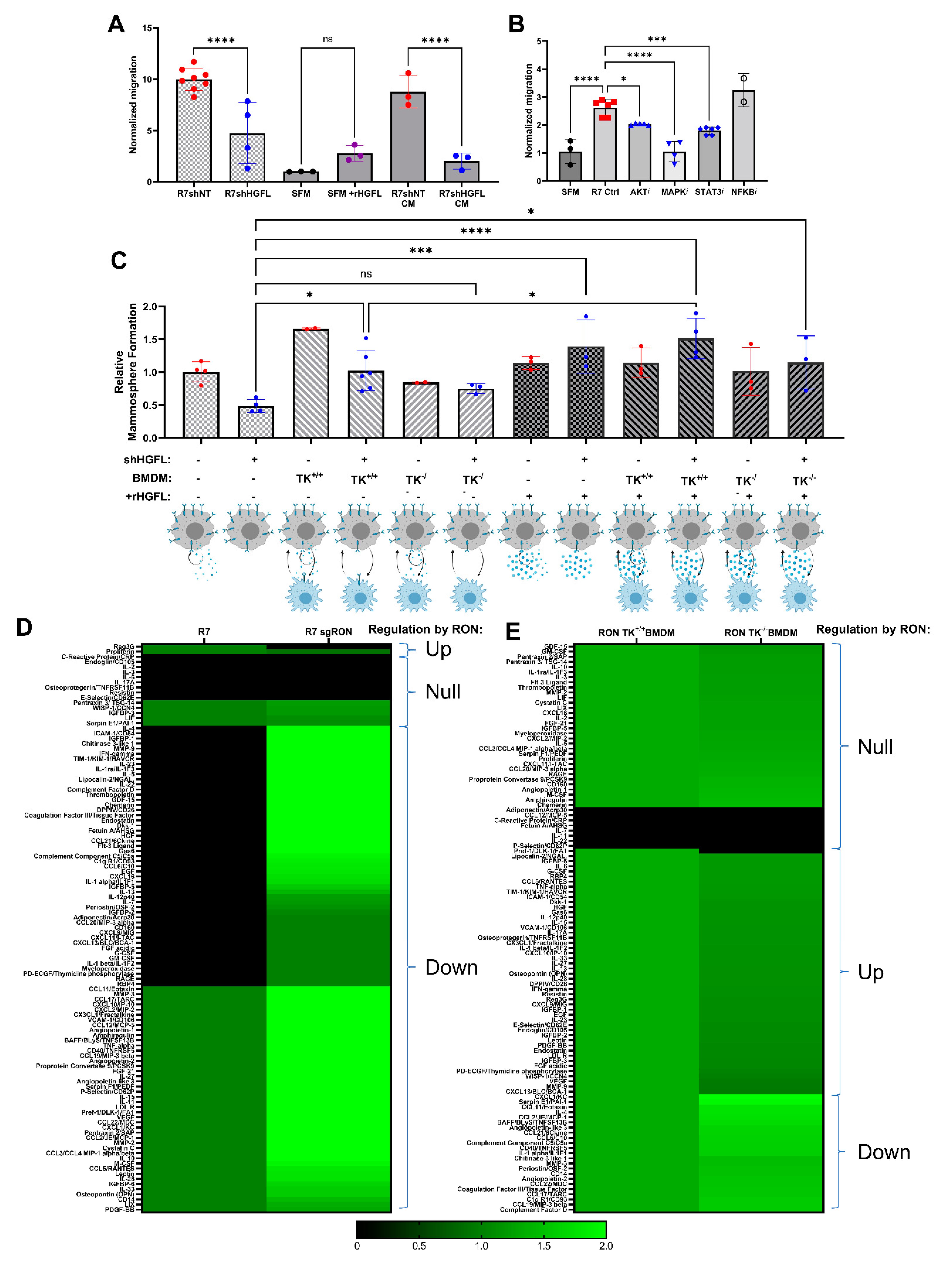
3.4. Tumor Cell-Secreted HGFL Promotes Macrophage Migration and Mammosphere Formation through Autocrine and Paracrine Mechanisms
3.5. RON Signaling Induces Changes to the Secretome of Tumor Cells and Macrophages
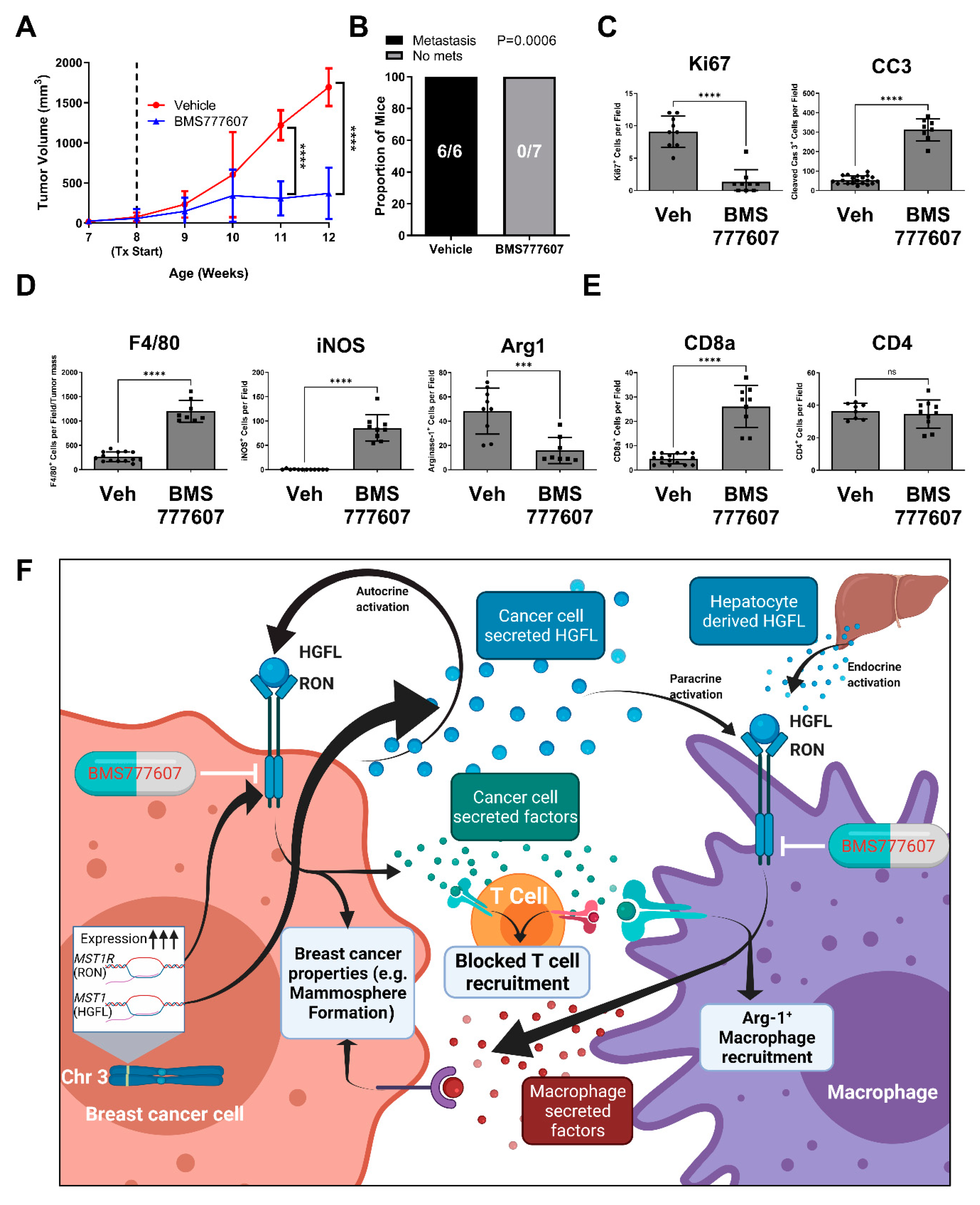
3.6. Inhibiting HGFL-RON Signaling in Tumor Cells and Macrophages via BMS777607 Impairs Tumor Growth, Metastasis, and Tumoral Macrophage Recruitment
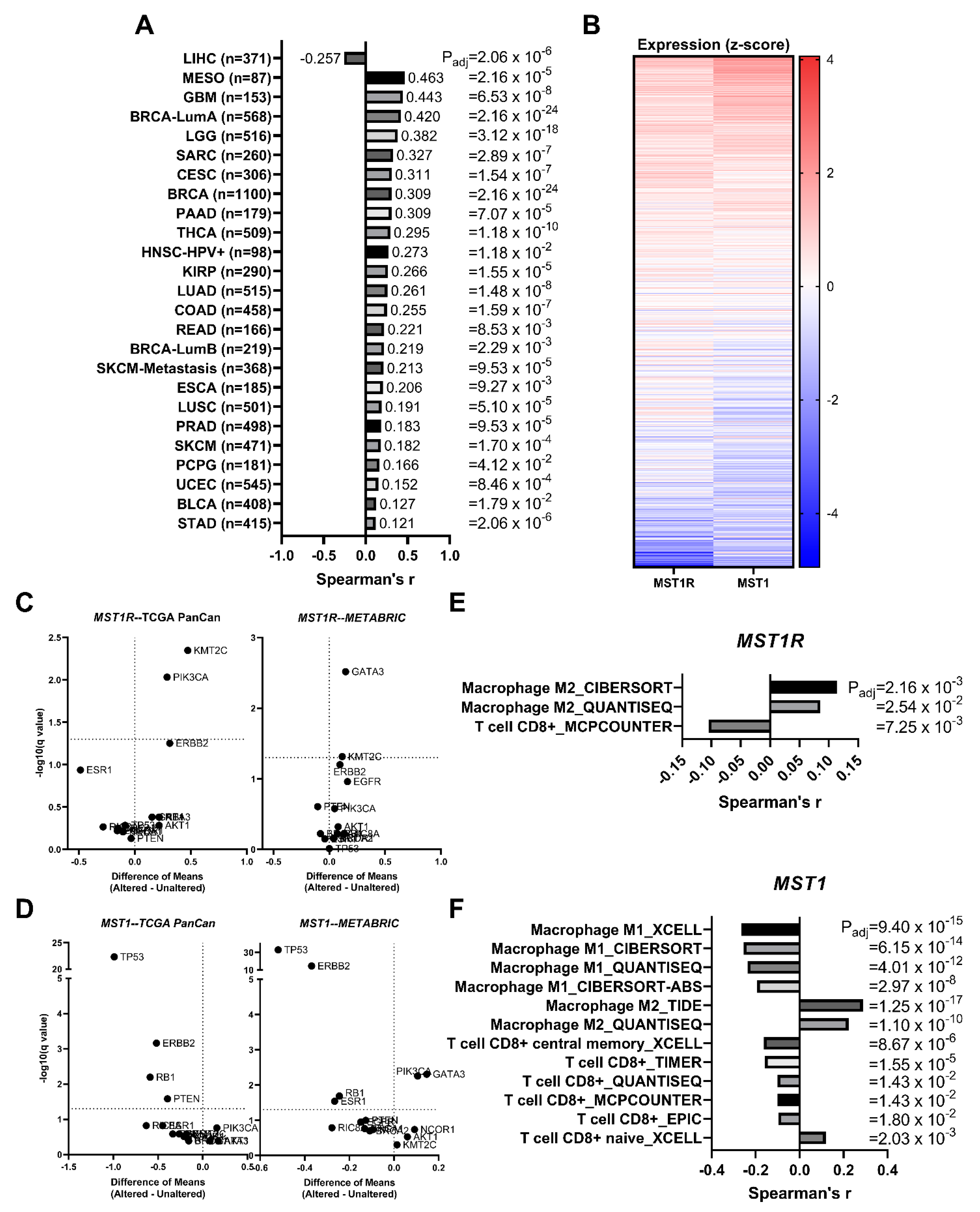
3.7. MST1R (RON) and MST1 (HGFL) Show Correlated Gene Expression in Virtually All Tumor Types including Breast Cancer
3.8. MST1R and MST1 Genes Are Virtually Never Altered/Mutated in Breast Cancer, and MST1R/MST1 Expression Is Not Associated with Any Recurrent Breast Cancer Driver Alterations/Mutations
3.9. MST1R and MST1 Genes Are Correlated with M2 Macrophage Infiltration Gene Signatures and Anti-Correlated with CD8+ T Cell Infiltration Gene Signatures in Human Breast Cancer Patient Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mallakin, A.; Kutcher, L.W.; McDowell, S.A.; Kong, S.; Schuster, R.; Lentsch, A.B.; Aronow, B.J.; Leikauf, G.D.; Waltz, S.E. Gene expression profiles of Mst1r-deficient mice during nickel-induced acute lung injury. Am. J. Respir. Cell Mol. Biol. 2006, 34, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, W.D.; Kulkarni, R.M.; Gray, J.K.; Vasiliauskas, J.; Leonis, M.A.; Waltz, S.E. Ron receptor regulates Kupffer cell-dependent cytokine production and hepatocyte survival following endotoxin exposure in mice. Hepatology 2011, 53, 1618–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Chanda, D.; Shiri-Sverdlov, R.; Neumann, D. MSP: An emerging player in metabolic syndrome. Cytokine Growth Factor Rev. 2015, 26, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Zinser, G.M.; Leonis, M.A.; Toney, K.; Pathrose, P.; Thobe, M.; Kader, S.A.; Peace, B.E.; Beauman, S.R.; Collins, M.H.; Waltz, S.E. Mammary-specific Ron receptor overexpression induces highly metastatic mammary tumors associated with beta-catenin activation. Cancer Res. 2006, 66, 11967–11974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benight, N.M.; Wagh, P.K.; Zinser, G.M.; Peace, B.E.; Stuart, W.D.; Vasiliauskas, J.; Pathrose, P.; Starnes, S.L.; Waltz, S.E. HGFL supports mammary tumorigenesis by enhancing tumor cell intrinsic survival and influencing macrophage and T-cell responses. Oncotarget 2015, 6, 17445–17461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Torres, S.J.; Benight, N.M.; Karns, R.A.; Lower, E.E.; Guan, J.L.; Waltz, S.E. HGFL-mediated RON signaling supports breast cancer stem cell phenotypes via activation of non-canonical beta-catenin signaling. Oncotarget 2017, 8, 58918–58933. [Google Scholar] [CrossRef] [Green Version]
- Bourn, J.R.; Ruiz-Torres, S.J.; Hunt, B.G.; Benight, N.M.; Waltz, S.E. Tumor cell intrinsic RON signaling suppresses innate immune responses in breast cancer through inhibition of IRAK4 signaling. Cancer Lett. 2021, 503, 75–90. [Google Scholar] [CrossRef]
- Eyob, H.; Ekiz, H.A.; DeRose, Y.S.; Waltz, S.E.; Williams, M.A.; Welm, A.L. Inhibition of Ron kinase blocks conversion of micrometastases to overt metastases by boosting anti-tumor immunity. Cancer Discov. 2013, 3, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Babicky, M.L.; Harper, M.M.; Chakedis, J.; Cazes, A.; Mose, E.S.; Jaquish, D.V.; French, R.P.; Childers, B.; Alakus, H.; Schmid, M.C.; et al. MST1R kinase accelerates pancreatic cancer progression via effects on both epithelial cells and macrophages. Oncogene 2019, 38, 5599–5611. [Google Scholar] [CrossRef]
- Wagh, P.K.; Peace, B.E.; Waltz, S.E. Met-related receptor tyrosine kinase Ron in tumor growth and metastasis. Adv. Cancer Res. 2008, 100, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Gurusamy, D.; Ruiz-Torres, S.J.; Johnson, A.L.; Smith, D.A.; Waltz, S.E. Hepatocyte growth factor-like protein is a positive regulator of early mammary gland ductal morphogenesis. Mech. Dev. 2014, 133, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.P.; Zhou, Y.Q.; Zhang, R.; Wang, M.H. MSP-RON signalling in cancer: Pathogenesis and therapeutic potential. Nat. Rev. Cancer 2013, 13, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, P.; Marchio, S.; Stella, M.C.; Giai, M.; Belfiore, A.; De Bortoli, M.; Di Renzo, M.F.; Costantino, A.; Sismondi, P.; Comoglio, P.M. Overexpression of the RON gene in human breast carcinoma. Oncogene 1998, 16, 2927–2933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, B.G.; Wicker, C.A.; Bourn, J.R.; Lower, E.E.; Takiar, V.; Waltz, S.E. MST1R (RON) expression is a novel prognostic biomarker for metastatic progression in breast cancer patients. Breast Cancer Res. Treat. 2020, 181, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Chen, H.H.; Chow, N.H.; Su, W.C.; Lin, P.W.; Guo, H.R. Prognostic significance of co-expression of RON and MET receptors in node-negative breast cancer patients. Clin. Cancer Res. 2005, 11, 2222–2228. [Google Scholar] [CrossRef] [Green Version]
- Peace, B.E.; Toney-Earley, K.; Collins, M.H.; Waltz, S.E. Ron receptor signaling augments mammary tumor formation and metastasis in a murine model of breast cancer. Cancer Res. 2005, 65, 1285–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Torres, S.J.; Bourn, J.R.; Benight, N.M.; Hunt, B.G.; Lester, C.; Waltz, S.E. Macrophage-mediated RON signaling supports breast cancer growth and progression through modulation of IL-35. Oncogene 2022, 41, 321–333. [Google Scholar] [CrossRef]
- Bezerra, J.A.; Witte, D.P.; Aronow, B.J.; Degen, S.J. Hepatocyte-specific expression of the mouse hepatocyte growth factor-like protein. Hepatology 1993, 18, 394–399. [Google Scholar]
- Vasiliauskas, J.; Nashu, M.A.; Pathrose, P.; Starnes, S.L.; Waltz, S.E. Hepatocyte growth factor-like protein is required for prostate tumor growth in the TRAMP mouse model. Oncotarget 2014, 5, 5547–5558. [Google Scholar] [CrossRef] [Green Version]
- Fluck, M.M.; Schaffhausen, B.S. Lessons in signaling and tumorigenesis from polyomavirus middle T antigen. Microbiol. Mol. Biol. Rev. 2009, 73, 542–563. [Google Scholar] [CrossRef] [Green Version]
- Regua, A.T.; Arrigo, A.; Doheny, D.; Wong, G.L.; Lo, H.W. Transgenic mouse models of breast cancer. Cancer Lett. 2021, 516, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Gurusamy, D.; Gray, J.K.; Pathrose, P.; Kulkarni, R.M.; Finkleman, F.D.; Waltz, S.E. Myeloid-specific expression of Ron receptor kinase promotes prostate tumor growth. Cancer Res. 2013, 73, 1752–1763. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.E.; Paluch, A.M.; Nashu, M.A.; Komurov, K.; Waltz, S.E. Tumor Cell Autonomous RON Receptor Expression Promotes Prostate Cancer Growth Under Conditions of Androgen Deprivation. Neoplasia 2018, 20, 917–929. [Google Scholar] [CrossRef]
- Kocher, T.; Asslaber, D.; Zaborsky, N.; Flenady, S.; Denk, U.; Reinthaler, P.; Ablinger, M.; Geisberger, R.; Bauer, J.W.; Seiffert, M.; et al. CD4+ T cells, but not non-classical monocytes, are dispensable for the development of chronic lymphocytic leukemia in the TCL1-tg murine model. Leukemia 2016, 30, 1409–1413. [Google Scholar] [CrossRef] [Green Version]
- Faustino-Rocha, A.; Oliveira, P.A.; Pinho-Oliveira, J.; Teixeira-Guedes, C.; Soares-Maia, R.; da Costa, R.G.; Colaco, B.; Pires, M.J.; Colaco, J.; Ferreira, R.; et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab. Anim. 2013, 42, 217–224. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Kulkarni, R.M.; Stuart, W.D.; Gurusamy, D.; Waltz, S.E. Ron receptor signaling is protective against DSS-induced colitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G1065–G1074. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Teitelbaum, S.L.; Zou, W.; Zheng, Y.; Johnson, J.F.; Chappel, J.; Ross, F.P.; Zhao, H. Cdc42 regulates bone modeling and remodeling in mice by modulating RANKL/M-CSF signaling and osteoclast polarization. J. Clin. Investig. 2010, 120, 1981–1993. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- cBioPortal. Available online: https://www.cbioportal.org/ (accessed on 12 July 2021).
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Hua, L.; Harmer, D.; Li, P.; Ren, G. Cre Driver Mice Targeting Macrophages; Springer: New York, NY, USA, 2018; pp. 263–275. [Google Scholar]
- Nikolaidis, N.M.; Kulkarni, R.M.; Gray, J.K.; Collins, M.H.; Waltz, S.E. Ron receptor deficient alveolar myeloid cells exacerbate LPS-induced acute lung injury in the murine lung. Innate Immun. 2011, 17, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Andrade, K.; Fornetti, J.; Zhao, L.; Miller, S.C.; Randall, R.L.; Anderson, N.; Waltz, S.E.; McHale, M.; Welm, A.L. RON kinase: A target for treatment of cancer-induced bone destruction and osteoporosis. Sci. Transl. Med. 2017, 9, eaai9338. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Daa, T.; Yada, N.; Kashima, K.; Fujitomi, Y.; Yokoyama, S. Expression and mutational status of RON in neoplastic lesions of the breast: Analysis of MSP/RON signaling in ductal carcinoma in situ and invasive ductal carcinoma. APMIS 2012, 120, 358–367. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Queried Datasets | Number of Samples with Alterations in Specified Gene(s) | Statistics | ||||||
|---|---|---|---|---|---|---|---|---|
| MST1R | MST1 | Both | Neither | p-Value | Q-Value | Log2 Odds Ratio | Tendency | |
| TCGA PanCan, MBC Provisional, MBC INSERM | 7 | 5 | 10 | 1483 | <0.001 | <0.001 | >3 | Co-occurrence |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hunt, B.G.; Jones, A.; Lester, C.; Davis, J.C.; Benight, N.M.; Waltz, S.E. RON (MST1R) and HGFL (MST1) Co-Overexpression Supports Breast Tumorigenesis through Autocrine and Paracrine Cellular Crosstalk. Cancers 2022, 14, 2493. https://doi.org/10.3390/cancers14102493
Hunt BG, Jones A, Lester C, Davis JC, Benight NM, Waltz SE. RON (MST1R) and HGFL (MST1) Co-Overexpression Supports Breast Tumorigenesis through Autocrine and Paracrine Cellular Crosstalk. Cancers. 2022; 14(10):2493. https://doi.org/10.3390/cancers14102493
Chicago/Turabian StyleHunt, Brian G., Angelle Jones, Carissa Lester, James C. Davis, Nancy M. Benight, and Susan E. Waltz. 2022. "RON (MST1R) and HGFL (MST1) Co-Overexpression Supports Breast Tumorigenesis through Autocrine and Paracrine Cellular Crosstalk" Cancers 14, no. 10: 2493. https://doi.org/10.3390/cancers14102493
APA StyleHunt, B. G., Jones, A., Lester, C., Davis, J. C., Benight, N. M., & Waltz, S. E. (2022). RON (MST1R) and HGFL (MST1) Co-Overexpression Supports Breast Tumorigenesis through Autocrine and Paracrine Cellular Crosstalk. Cancers, 14(10), 2493. https://doi.org/10.3390/cancers14102493