Simple Summary
Biliary tract cancers (BTCs) are a rare but deadly group of gastrointestinal tumors that are often diagnosed in the advanced stages of disease. Despite large studies investigating optimal systemic therapy options in BTCs, current chemotherapies provide only modest benefits in overall survival. The rapidly evolving study of homologous recombination repair (HRR) as part of the broader DNA damage repair (DDR) system makes it possible to exploit deficiencies in this pathway with targeted agents such as PARP inhibitors (PARPi). We will review the rationale for PARPi use, as well as considerations for further unlocking their potential in treating BTC.
Abstract
Biliary tract cancers (BTCs) are a heterogeneous group of malignancies that make up ~7% of all gastrointestinal tumors. It is notably aggressive and difficult to treat; in fact, >70% of patients with BTC are diagnosed at an advanced, unresectable stage and are not amenable to curative therapy. For these patients, chemotherapy has been the mainstay treatment, providing an inadequate overall survival of less than one year. Despite the boom in targeted therapies over the past decade, only a few targeted agents have been approved in BTCs (i.e., IDH1 and FGFR inhibitors), perhaps in part due to its relatively low incidence. This review will explore current data on PARP inhibitors (PARPi) used in homologous recombination deficiency (HRD), particularly with respect to BTCs. Greater than 28% of BTC cases harbor mutations in genes involved in homologous recombination repair (HRR). We will summarize the mechanisms for PARPi and its role in synthetic lethality and describe select genes in the HRR pathway contributing to HRD. We will provide our rationale for expanding patient eligibility for PARPi use based on literature and anecdotal evidence pertaining to mutations in HRR genes, such as RAD51C, and the potential use of reliable surrogate markers of HRD.
1. Introduction
Biliary tract cancers (BTCs) arise from bile duct epithelia within the liver (intrahepatic) and outside the liver (extrahepatic). They are heterogeneous and constitute ~7% of all gastrointestinal (GI) tumors [1]. In the Western world, BTC occurs at a rate of 0.5 to 2 cases per 100,000 people [2]. BTCs tend to be aggressive, with a 5-year OS of <10% [3]. Surgery is the only means of a cure; however, upwards of 70% of patients are diagnosed with advanced disease that is unresectable [2,4,5]. Studies found that patient median disease-free survival (DFS) was 12–36 months after surgical resection, and their overall survival (OS) was high as 80 months after R0 resection [6,7,8]. Still, disease recurrence after curative surgery remains an issue, and recurrence rates are as high as 40–70% [9,10,11].
Until the last couple of years, the standard treatment for BTCs was chemotherapy with palliative intent. Gemcitabine plus cisplatin (GEMCIS) is still the most commonly used front-line regimen in the United States, and fluorouracil plus oxaliplatin (FOLFOX) is the typical second-line treatment, supported by the Phase III ABC-02 trial and the ABC-06 trial, respectively [12,13]. Nonetheless, survival of patients with advanced BTCs remains abysmal, with mOS being under one year with traditional chemotherapy [14].
Over the past decade, breakthrough therapies in BTC have revolved around targeted therapies and immunotherapies. Increased use of broad molecular profiling has led to the finding that BTCs have some of the highest genetic aberration rates of all cancers. Studies have shown that upwards of 60% of cholangiocarcinoma (CCA) contain such aberrations, including those in isocitrate dehydrogenase-1 (IDH1), fibroblast growth factor receptor-2 (FGFR2), and neurotrophic tyrosine receptor kinase (NTRK) [15,16,17,18]. Accordingly, molecularly targeted agents have been developed and studied in BTC, leading to the recent United States Food and Drug Administration (USFDA) approval of ivosidenib (IDH1 inhibitor) and pemigatinib (FGFR 1,2,3 inhibitors) [19,20].
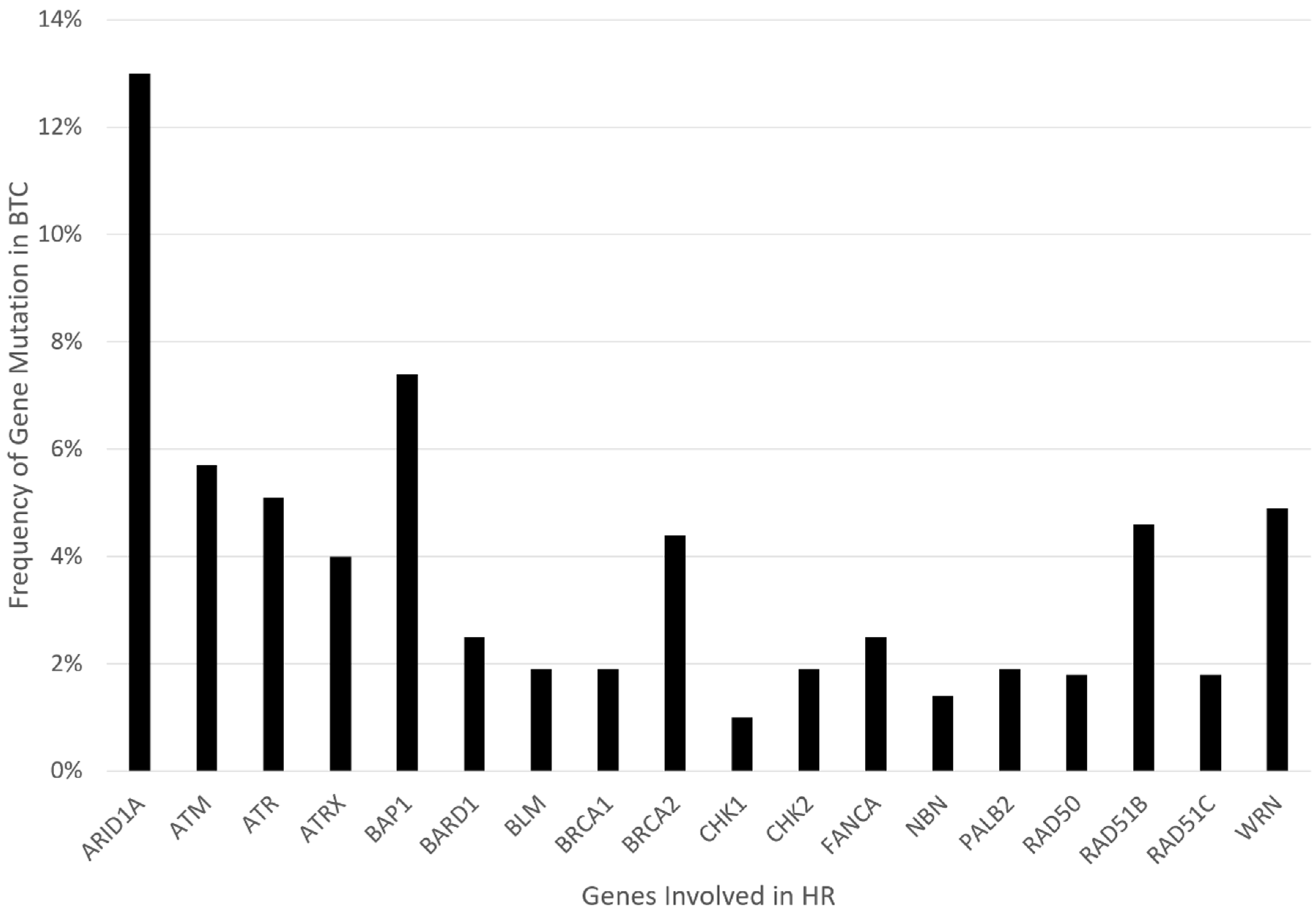
Similar to the mutations named above, the focus of our review is on the role of poly (ADP-ribose) polymerase inhibitors (PARPi) in homologous recombination repair (HRR) deficient tumors, particularly concerning BTCs. This mainly encompasses genetic alterations that are linked to genes in the HR pathway, including but not limited to ARID1A, ATM, ATRX, BAP1, BARD1, BLM, BRCA1/2, BRIP1, CHEK1/2, FANCA/C/D2/E/F/G/L, MRE11A, NBN, PALB2, RAD50, RAD51, or WRN. Retrospective gene analysis found evidence of HRR deficiency (HRD) in 28.9% of BTCs, one of the highest HRD rates of all cancers [21,22,23]. Figure 1 presents the relative frequency data for these gene mutations in BTC, extracted from the COSMIC/TCGA database. PARP inhibitors (PARPi) are the first group of drugs designed to exploit cancer HRD. Preclinical studies suggest that BTC is susceptible to PARP inhibition. Several PARPi, including olaparib, rucaparib, and niraparib, are under clinical investigation in BTCs [24]. In this review, we will discuss the predictors of PARPi sensitivity and resistance in both clinical and preclinical studies. Furthermore, synergistic strategies through combination therapy with other drugs will be addressed.
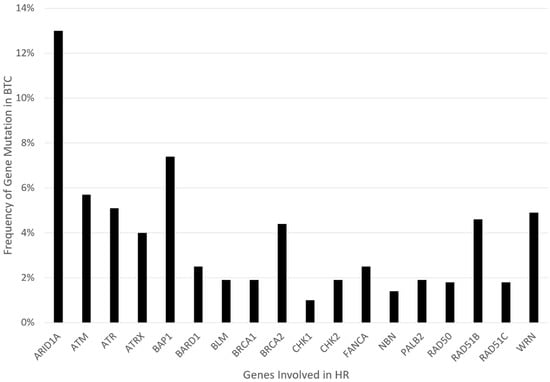
Figure 1.
Graphical representation of mutation frequency in genes involved in HR.
2. The DNA Damage Repair Pathway
Normal cells are under constant stress from external and internal factors, which can cause cell and DNA damage. Cells rely on intricate DNA damage repair (DDR) pathways to identify and fix errors to ensure genomic integrity. Various DNA repair mechanisms exist to address an array of different mutations and alterations. The major DNA repair pathways include (but are not limited to) base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), non-homologous end joining (NHEJ), and homologous recombination repair (HRR). For example, BER has activity in repairing single-stranded breaks (SSB), while double-stranded breaks (DSB), often highly cytotoxic if not repaired correctly, are typically fixed through error-proof HRR or more error-prone classical NHEJ and alternative-NHEJ pathways such as microhomology-mediated end joining (MMEJ) [25,26]. Specifically, HRR is active during the S and G2 phase of the cell cycle using a sister chromatid template for DSB repair and is also involved in replication forks that have stalled or collapsed [26,27].
In response to DNA damage, these main DDR routes operate under a dynamic and complex network of overlapping signaling pathways that crosstalk and either promote genomic stability or trigger programmed cell death, as deemed appropriate under the circumstances [28]. DDR defects theoretically provide a number of advantages during carcinogenesis; an increased mutation rate helps tumors withstand replicative stress and offers an adaptive advantage. Mutations in DDR genes have been identified in many cancers, and it has been proposed that every DDR mechanism is affected during tumorigenesis [29,30]. However, the impact of many genes in the DDR pathway is not yet well understood. Herein, we will discuss the evidence surrounding the BRCA-mutant phenotype in some BRCA-wild type tumors and the need for additional biomarkers to detect this phenotype. Table 1 summarizes a non-exhaustive list of HRD-related genes, their frequencies in CCA, and the evidence surrounding their sensitivity to DDR-targeting agents.

Table 1.
Genes involved in HR, their frequencies in BTC, and a summary of evidence in support of sensitivity to PARPi when mutated.
3. Biomarkers of HRD and Predictors of PARPi Response in BTC
PARPi are the first and only class of drugs approved clinically to target DDR deficiency. PARP proteins are essential to DNA repair in a compensatory pathway to HRR that facilitates the repair of SSBs. PARPi have cytotoxic properties via two mechanisms: (1) they inhibit the enzymatic activity of PARP1 and PARP2 by binding to their cofactor β-NDA+ binding site, and (2) they “trap” PARP1 and thereby prevent the release of PARP1 from damaged DNA, leading to cell death in dsDNA repair-deficient cells [73]. PARP1 was initially described as a key facilitator of BER in ssDNA repair, although there is now increasing evidence that it also has direct roles in non-HRR pathways such as NHEJ [74,75,76]. PARPi efficacy in cancers with HRD exploits the concept of synthetic lethality. Synthetic lethality occurs when two (or more) DDR pathways are blocked, preventing effective DNA repair [74]. For example, if a BRCA mutation renders HRR defective, pharmacologic inhibition of PARP enables synthetic lethality by blocking at least two other repair pathways.
Although PARPi were initially studied and approved in BRCA1/2 mutated ovarian cancers, its use has been expanded to other cancers, including breast cancer, pancreatic cancer, and prostate cancer. Currently, not a single PARPi has been approved by the USFDA for BTC, although several trials are investigating PARPi use in BTC, and published data are pending. Therefore, much of this discussion is supported by data extrapolated from other cancers and preclinical models while being cautious of the biological differences between different tumor types.
When considering predictors of response to PARPi, BRCA 1 and 2 represent the iconic gene mutations that disrupt HRR and increase the sensitivity of cancer cells to PARPi. The phase III PAOLA trial impressively reported improved mOS of 37.2 months vs. 17.7 months (HR 0.33, 95% CI 0.25–0.45), with the addition of olaparib to bevacizumab vs. bevacizumab alone in stage III/IV BRCA mutated ovarian cancer in the first-line maintenance setting [61]. However, the investigators noted that in patients with HRD-positive tumors without BRCA mutations, the mOS was still much improved with the addition of olaparib (28.1 months vs. 15.5 months, HR 0.43, CI 95% 0.28–0.66). The SOLO3 trial showed that treatment with olaparib in platinum-sensitive, recurrent BRCA-positive ovarian cancer yielded an objective response rate (ORR) of 72% vs. 51% with nonplatinum chemotherapy [77].
The DDR pathway is complex; however, it potentially bears many opportunities for targeted therapies. In fact, as previously alluded to, varying degrees of BRCA-mutated phenotype, or “BRCAness”, are evident across many cancers that are BRCA-wild type or have BRCA mutations of unclear significance. With particular relevance to BTC, we propose two goals for this review: (1) exploring the qualifications for patients who may benefit from DDR targeting drugs such as PARPi beyond just BRCA mutations and (2) exploring the mechanisms of PARPi refractoriness, resistance, ability, and potential ways to overcome this.
As mentioned earlier, the PAOLA trial for ovarian cancer demonstrated significant benefit from olaparib for patients who were HRD-positive and BRCA-negative. In BTC, while only 3–4% of tumors cary BRCA mutations, as high as 28.9% of tumors carry mutations in genes involved in the HRR pathway [21,78]. Hence, we seek to characterize a group of patients with BRCA wild type BTC who may benefit from DDR targeting therapies such as PARPi.
4. ARID1A
To explore a few common DDR mutations in BTC, the most commonly detected gene alteration is ARID1A (AT-rich interactive domain-containing protein 1A), occurring in 13–15% of BTC samples [21,22,23]. Most ARID1A mutations are truncating mutations (92%), leading to the gene’s inactivation and loss of its protein expression [79]. The clinicopathologic features of ARID1A are somewhat controversial. Most studies suggest a tumor suppressor role for this gene, whereas others describe pro-oncogenic properties [79]. Its oncogenic properties are not well understood and are seemingly complex; ARID1A appears to increase oxidative stress, such as by increasing CYP450 in the liver in hepatocellular carcinoma (HCC). It is overexpressed in primary HCC but, interestingly, lost in metastatic HCC [80].
As the most frequently mutated chromatin regulator across all cancers, it is important to note the role of ARID1A in global transcription regulation [81]. Specifically, depletion of ARID1A leads to decreased RNA polymerase II (RNAPII) pausing. The pausing of RNAPII is essential for maintaining cell homeostasis in response to environmental stimuli and limiting excessive transcription [81]. Furthermore, co-occurring TP53 and ARID1A mutations are rare; this mutual exclusivity has been demonstrated in endometrial, gastric, breast, and esophageal cancers [82]. Studies found that some, but not all, transcriptional dysregulation suffered from ARID1A loss can be rescued by ARID1B and TP53 targets [81]. In fact, the rare coexistence of TP53 and ARID1A mutations often leads to more aggressive cancers [82].
When considering its role in DDR, ARID1A exerts its role in both HR and NHEJ pathways. It is also required for proper G2/M DNA damage checkpoint, which may lead to insufficient cell cycle arrest to repair DSB [31]. Its deficiency has been demonstrated to impair the DSB end resection necessary for repair. ARID1A is a member of the SWI/SNF complex and is involved in chromatin remodeling, which promotes efficient DSB end resection and sustains ATP-dependent signaling [31,32]. The aberrancies in the SWI/SNF complex as a whole have been linked to various cancers and linked to tumor suppression. Interestingly, ARID1A and TP53 are often found to be co-mutated in CCA, which would indicate a more aggressive cancer given the collaboration of these two genes in the activation of downstream effectors to prevent tumorigenesis, as noted above [83,84].
In preclinical studies, cell lines with knockout ARID1A demonstrated sensitivity to PARPi. Moreover, selective inhibition of ARID1A-deficient xenografts by PARPi was observed [31]. These findings have not been validated clinically. On the contrary, retrospective analysis of data from the ARIEL2 trial identified ARID1A persistence as a factor of rucaparib resistance. He and colleagues found that all 10 ovarian cancer patients with loss of ARID1A and BRCA-wild type from the study had significantly less PFS on rucaparib than proficient ARID1A cancers [34]. Nonetheless, there is insufficient data to delineate the mechanism of such resistance or suggest the clinical prognostic or predictive role of ARID1A in BTC. Further prospective clinical trials are necessary to evaluate PARPi efficacy in ARID1A mutated cancers, particularly given its relatively high frequency across all tumor types.
5. ATM and ATR
ATM (Ataxia-telangiectasia mutated) and ATR (ATM- and Rad3-related) are part of the PI3K-related kinase family. When mutated, they are associated with complex syndromes that exhibit nervous system phenotypes and DDR deficiencies [85]. ATM and ATR mutations are also among the most common associated HRD mutations in BTC, occurring in 5.7% and 5.1% of BTC tumor samples, respectively [22,23]. ATM functions upstream in the HRR pathway, and both help phosphorylate downstream HRR effectors, including BRCA1. ATM and ATR show complementary relationships, likely through compensatory mechanisms, such that ATM knockout cells have increased levels of ATR [85]. Hence, there is the suggestion that the two genes work together in the DDR pathway, and it would be reasonable to consider this as a mechanism of resistance to DDR targeting agents in ATM or ATR deficient cells.
It has been shown that patients with ATM-deficient tumors are more responsive to radiotherapy, platinum-based chemotherapy, and PARPi [35]. Similarly, in ovarian cancer xenografts, a group showed that administering an ATR inhibitor helped overcome platinum resistance [41]. A phase II double-blind trial of olaparib plus paclitaxel vs. paclitaxel alone showed significant improvement in mOS (13.1 v 8.3 months, HR 0.35, p = 0.002) for patients with recurrent or metastatic gastric cancer with low ATM expression on immunostaining [86]. Given this clinical success, efforts are made to study ATM inhibitors, which artificially block normal ATM function. Several ATM inhibitors have entered early clinical phases in combination with cytotoxic therapies, PARPi, and/or radiation [NCT02588105, NCT03423628, NCT03571438].
6. IDH1
IDH1 mutations have a frequency of 20–25% in intrahepatic CCA. They indicate that BRCAness may not be exclusive to genes within the DDR pathway. Mutations in IDH1 across most tumors are predominantly somatic rather than germline mutations. They are always heterozygous, primarily because of the driver R132H mutation, consistent with gain of function and dominance over the remaining wild-type allele [87]. This mutant gene converts the α-ketoglutarate to 2-hydroxyglutarate (2-HG). 2-HG directly inhibits HRR, which was well captured by Sulkowski and colleagues [88]. In HCT116 and HeLa cell lines, these investigators first confirmed the existence of mutant IDH1 protein expression and a 100-fold increase in 2HG production. Compared to the IDH1-wild-type cell line, HCT116 and HeLa cell lines had a markedly reduced capacity for DSB repair after ionizing radiation exposure. Further testing via a plasmid reporter assay to compare relative DSB repair activity showed a marked deficiency in HRR in the mutant cells [88]. These mutational changes also sensitized the cell lines to further attack by PARPi. Although the exact mechanisms of its involvement in the HRR remain to be elucidated, mutant IDH1 was at least found to independently downregulate ATM in a mouse model [89].
In IDH1 mutated cancers, including BTC, PARP inhibitor efficacy is evident in preclinical studies but requires further clinical testing. IDH1 inhibitor ivosidenib has been approved by USFDA for CCA after demonstrating significantly improved PFS (6.9 months vs. 2.7 months in the placebo arm, p < 0.001) in a phase III trial. Although clinical data on PARPi use in IDH1 mutated CCA is lacking, olaparib is currently being tested in patients with advanced glioma or cholangiocarcinoma with IDH1/2 mutations [NCT03212274]. A phase II study of olaparib in IDH1/IDH2-mutant mesenchymal sarcoma also reported some signs of clinical benefit [90].
7. RAD52
Among several roles in DNA repair and replication, RAD52 binds single-stranded DNA and plays key roles in single-strand annealing and HRR of DSBs. In mammals, RAD52 has diminished HRR compared to that of other proteins, including BRCA1/2. In checkpoint-deficient cells, it facilitates break-induced replication (BIR), which is a specialized pathway that repairs single-ended DBS and promotes the finalization of DNA replication [66,67,68]. Interestingly, in the study of gene mutations across different malignancies, a RAD52 mutation was found to sensitize BRCA-deficient AML cells to PARPi. Deficiencies in KU70/80 proteins that compete with RAD52 in binding to DSB are related to resistance to PARPi, although mutations in KU70/80 have not been reported in BTC [91,92].
One of its most prominent functions is the formation of a Rad51–Rad52–Rad59 complex, which is involved in conservative recombination events, including gene conversion and reciprocal recombination [67]. RAD52-mediated DNA repair remains active in PARPi-treated BRCA-deficient tumor cells such that dual inhibition of RAD52 and PARP1 have demonstrated evidence of synthetic lethality in a BRCA-deficient mouse model [69]. It has other important functions in DNA repair, which are beyond the scope of the article.
8. RAD51 and RAD51C
The HRR genes of particular interest are RAD51 and its paralogs (e.g., RAD51C). During DSB end resection, 3′-tailed ends are created in the DNA and are quickly bound by replication protein A (RPA). RPA is then displaced by RAD51, which is central to a helical nucleoprotein filament that identifies and invades a homologous donor DNA duplex [93]. RAD51 is helped by other HRR proteins, most prominently BRCA2, which is recruited by BRCA1 and co-localizes with PALB2 at the site of DSB. When bound to RAD51, BRCA2 attenuates ATPase activity in RAD51 and assists in RAD51 filament formation and stabilization of the complex [93]. From there, RAD51 mediates homologous DNA strand identification and strand invasion to begin DNA replication in the recombination process.
Clinically, the correlation between RAD51 expression and tumorigenesis is complex. For example, low RAD51 expression correlated with high histologic grade in breast cancer but was predictive of higher complete pathologic response rates to neoadjuvant chemotherapy [94]. However, evidence also suggests that RAD51 expression is increased during breast cancer progression, and overexpression of RAD51 in colorectal cancer was a predictor of poor outcome [95,96]. During therapy with PARPi, immunostaining showed that increased expression of RAD51 nuclear foci correlated with PARPi resistance in BRCA mutated tumors [64]. Simultaneous targeting of RAD51 and PARP inhibition has been proposed as a synergistic approach, although there is currently no clinical data to confirm this proposal [94,96].
There are five paralogs of RAD51, including RAD51B, RAD51C, and RAD51D, which share similar functions in HRR. RAD51C mutations occur in 1.8% of BTC [22,23]. RAD51C colocalizes with RAD51 to DNA repair foci and also facilitates nucleoprotein filament stabilization. It participates in the formation of two complexes: the XRCC3-RAD51C (CX3) heterodimeric structure and RAD51B-RAD51C-RAD51D-XRCC2 (BCDX2) heterotetrameric structure [97]. Compared to the other RAD51 paralogs, this suggests that RAD51C plays a more prominent role in HRR given its unique involvement in both structures, which are central to the early and late stages of HR [98]. The CX3 and the BCDX2 complexes are also crucial for stalled replication fork reversal and efficient restart, for which HRR components are also generally implicated [99].
Germline RAD51C pathologic variants have been linked with an increased risk of tubo-ovarian carcinoma (RAD51C mutant RR 7.55, 95% CI = 5.60–10.19) and breast cancer (RAD51C mutant RR 1.99, 95% CI = 1.39–2.85) [100]. Clinically, immunostaining of high-grade serous ovarian cancer showed higher levels of RAD51C protein compared to benign tumors, and increased RAD51C levels were associated with higher clinical staging and poorer prognosis [97]. Given its integral role in HRR, studies suggest that RAD51C shares many tumor characteristics associated with BRCA germline variants [101]. In fact, in the ARIEL2 trial of the PARP inhibitor rucaparib in ovarian cancer, tumor biopsies revealed positive associations between alterations in BRCA1 or RAD51C, high genomic loss of heterozygosity (gLOH), and increased response to rucaparib [102]. Silencing of RAD51C expression has also been shown to make tumor cells more sensitive to PARPi in vitro and has also been replicated in clinical trials for patients with ovarian cancer who had an increased response rate to rucaparib [101,103]. Conversely, secondary mutations in RAD51C, which restore the function of RAD51C, are linked to the restoration of the HRR pathway and have been identified as a mechanism of PARPi resistance in xenograft models [104].
9. Genomic Loss of Heterozygosity
The last BRCAness marker we would like to discuss is the concept of gLOH, which is a measure of allelic imbalance when heterozygous somatic cells become homozygous due to the loss of one of the two alleles. In a retrospective study, biallelic BRCA1/2 alteration was associated with increased LoH across multiple cancer types, including biliary tract cancers (BTC), although the magnitude of this association was variable across each cancer type [105]. In BTC, the odds ratio for this association was 21.5. By plotting the sensitivity and specificity of classifying BRCA1/2 biallelic alteration vs. wild type using genomic LoH, the authors found a cutoff of >17.6% genomic LoH for BTC, which is very close to the >16% genomic LoH which was used in the ARIEL trial of rucaparib in ovarian cancer [106]. Although >17.6% may correspond to a BRCA-mutant phenotype, prospective clinical trials are necessary to evaluate varying levels of genomic LoH, including those with genomic LoH >17.6%, and the corresponding sensitivity to DDR-directed therapies, such as PARPi. The goal would be to find a gLOH cutoff representing a level of HRR deficiency that would sensitize BTC patients to such therapies in the hope that gLOH could become an umbrella biomarker that catches all clinical HRR deficient cancers.
10. Anecdotal Case of RAD51 Mutation in CCA
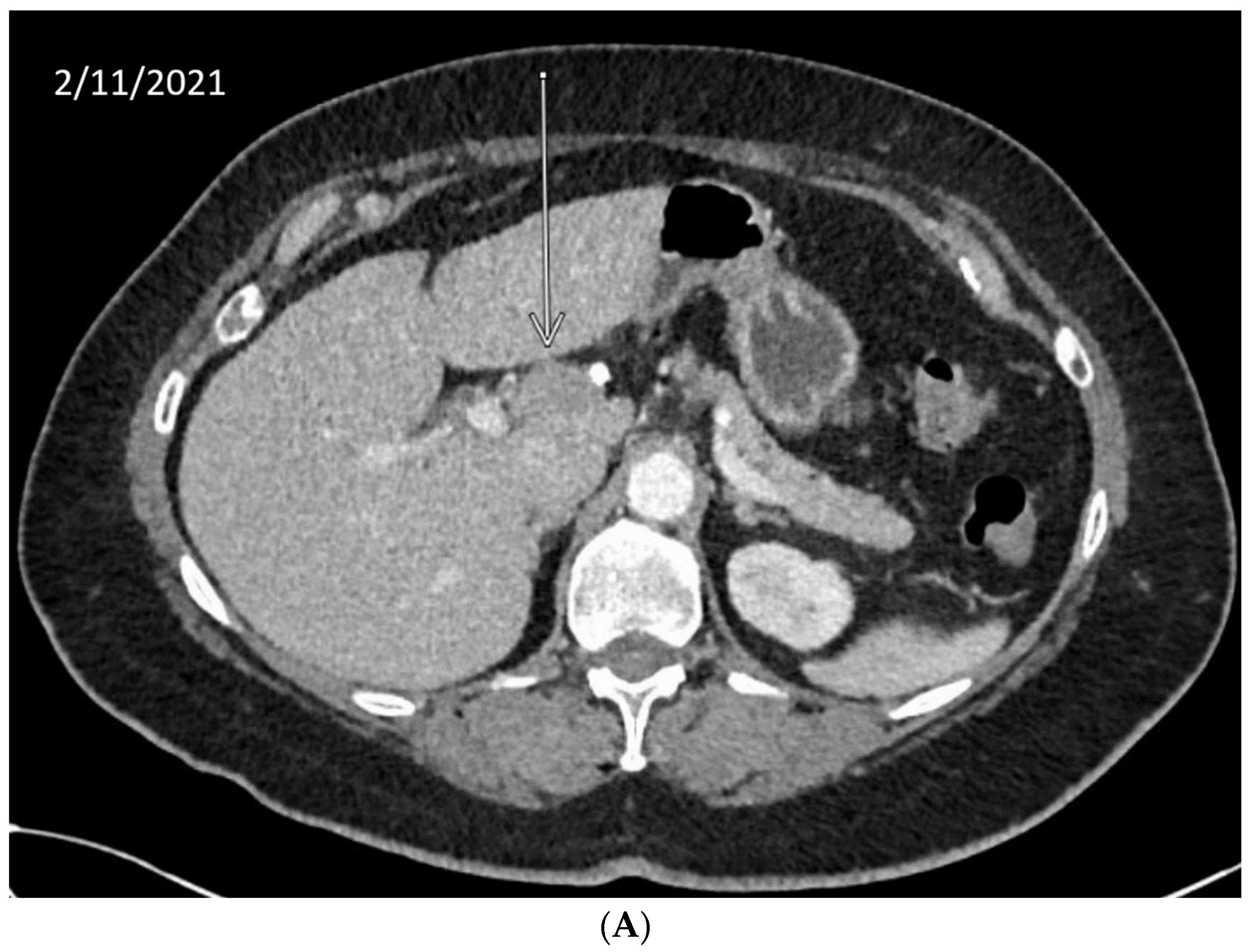
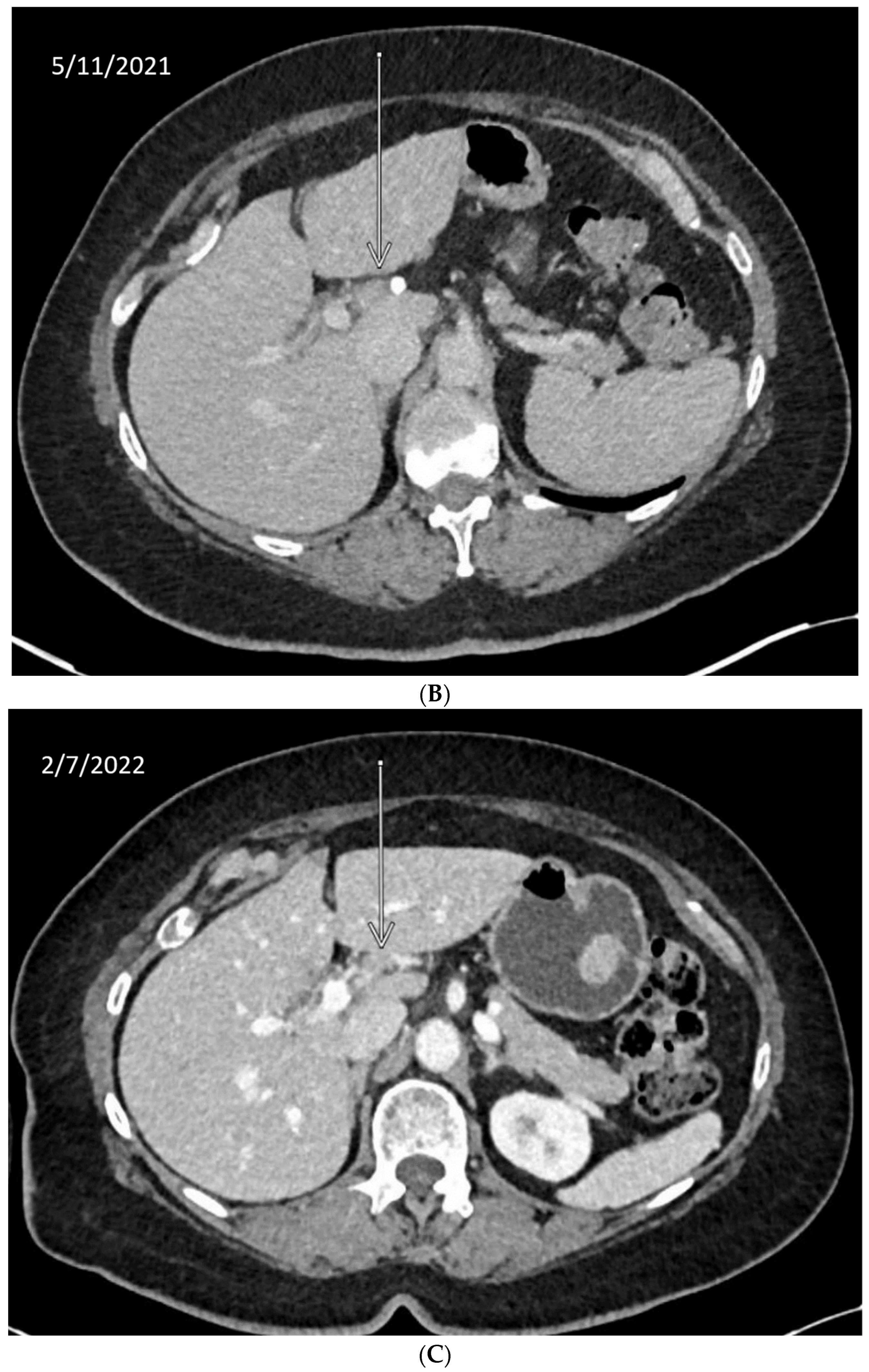
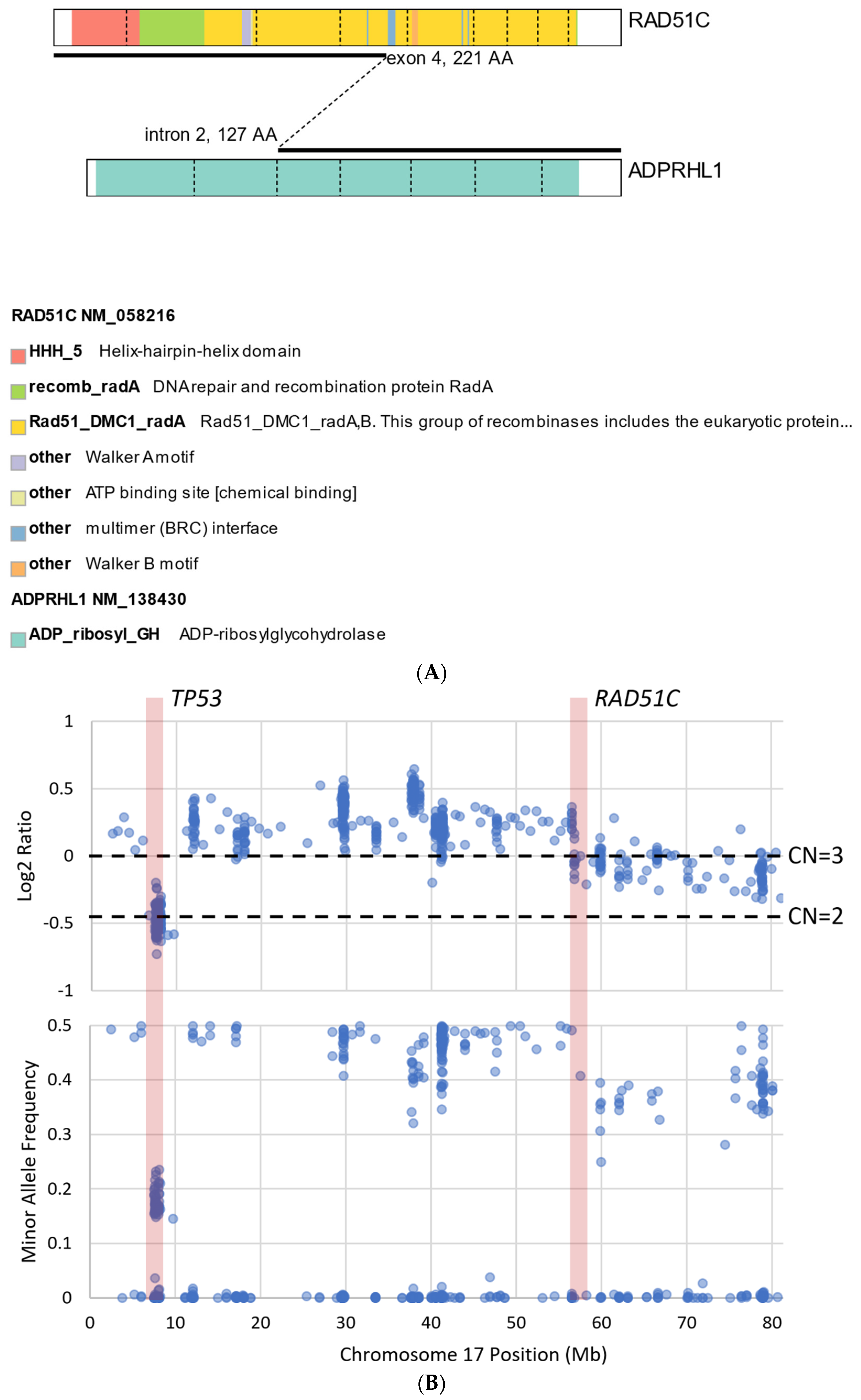
We describe our institutional experience of a 73-year-old woman presenting with metastatic BTC in March 2019. She was treated with GEMCIS between March and August 2019 and was switched to FOLFOX and fluorouracil maintenance therapy upon disease progression from August 2019 to December 2020, when she had further disease progression. She was then enrolled in our Phase II trial of olaparib plus pembrolizumab in relapsed/refractory advanced BTC in February 2021. As anticipated from her good response to platinum-based chemotherapy, the patient had an excellent partial response to PARPi in addition to ICI and a 65% decrease in index lesions (Figure 2). FoundationOne CDx comprehensive genomic profiling of the tumor showed a t(13;17) translocation with the chromosome 17 breakpoint identified in exon 4 of RAD51C (Figure 3A). This rearrangement had strong bi-directional evidence, with 237 supporting reads. Examination of chromosome 17 copy number data shows a copy number transition and accompanying LOH with a breakpoint at the RAD51C locus, suggesting likely loss of the second allele (Figure 3B). Genomic LOH, an orthogonal HRD signature validated as a companion diagnostic for rucaparib in ovarian cancer, was 11%, which is below the 16% threshold for gLOH-high status set in ovarian cancer. Biallelic inactivation of RAD51C has been associated with elevated gLOH [107]. However, the observed distribution of gLOH values varies by cancer type, and further study is needed to identify clinically relevant disease-specific thresholds [105]. The patient’s treatment was complicated by grade 3 immune hepatitis in June 2021; pembrolizumab has been held since then. She continued to respond to olaparib alone at the most recent clinical evaluation in February 2022.
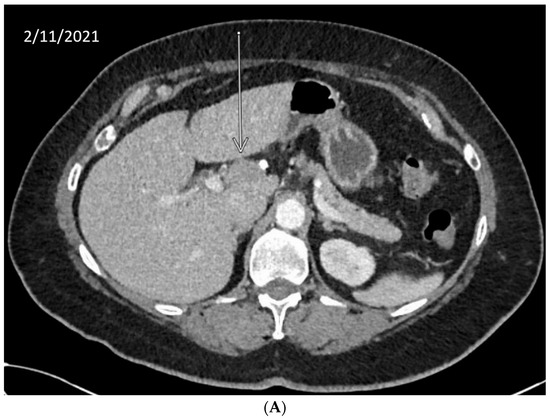
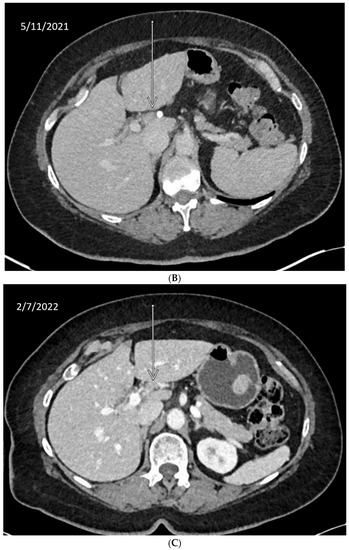
Figure 2.
Time-lapsed CT images of a patient with intrahepatic CCA and RAD51C mutation (via t(13;17) translocation with the chromosome 17 breakpoint identified in intron 4 of RAD51C). She concurrently had a genomic loss of heterozygosity (gLOH) of 11%. The patient had an excellent response to treatment with PARPi and ICI and a 65% decrease in index lesion (shown by arrow). (A) CT from February 11, 2021. (B) CT from May 11, 2021. (C) CT from 7 February 2022.
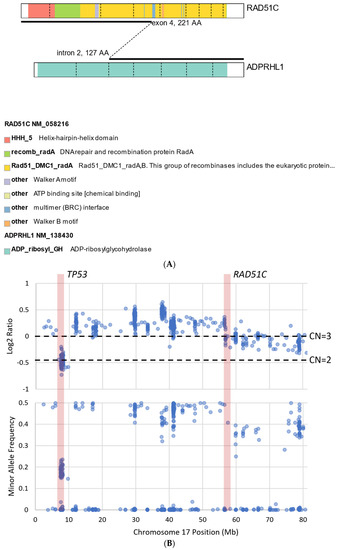
Figure 3.
Graphical illustration of RAD51C mutation from the same patient presented in Figure 2. (A) Translocation t(13;17) with chromosome 17 breakpoint identified in exon 4 of RAD51C. (B) Examination of chromosome 17 copy number data shows a copy number transition accompanying LOH with a breakpoint at the RAD51C locus, suggesting likely loss of the second allele.
11. Augmenting PARPi Efficacy
So far, we have explored aberrations in the HRR pathway that may suggest increased sensitivity to PARPi outside of the well-studied BRCA1/2 mutations. However, we turn our attention to ways to augment the efficacy of PARP inhibitors in BTC and other cancers.
First, we want to touch on the concept of drug resistance briefly. Tumor cells have the ability to develop ways to disrupt the availability of PARPi within the cell. For example, a high expression of ABCB1a/b, a drug-efflux transporter gene, has been associated with resistance to PARPi in mouse models [108]. ABCB1a/b overexpression is also a mechanism of resistance to topoisomerase inhibitors. However, by blocking the MDR1 glycoprotein encoded by ABCB1, researchers have demonstrated re-sensitization of ovarian cancer cell lines to PARPi. The co-administration of PARPi with MDR1 inhibitors, such as tariquidar and verapamil, or the development of PARPi that are non-substrates for MDR1-mediated efflux, is being studied [108].
The restoration of the HRR pathway in HRR-deficient cancers appears to occur by several different mechanisms. While this is sometimes achieved via compensatory mechanisms, such as increased activity of RAD52, studies have also demonstrated secondary somatic reversion of mutated genes in the DDR pathway, such as in BRCA1/2 and RAD51 genes, which can restore their function in cancer cells [68,109]. In high-grade ovarian cancers with germline BRCA1/2 mutations, almost 50% of platinum-resistant tumors demonstrated secondary mutations in the BRCA1/2 gene [109]. Several drug combination approaches have been proposed in these scenarios. For example, there is interest in CDK inhibition, given that CDK1 has a direct impact on BRCA1/2 activation through phosphorylation of BRCA1/2 [110]. PI3K inhibitors can also downregulate BRCA expression in cell lines and human-derived xenografts, and this downregulation is likely mediated by ERK signaling and impaired recruitment of RAD51 to DNA repair sites [110,111]. An early phase I clinical trial suggested synergy between alpelisib (PI3K inhibitor) and olaparib in ovarian cancer [112].
The idea of adding secondary agents to combat PARPi resistance or increase efficacy segues into another fascinating area of drug synergy: PARP plus PD-1/PD-L1 inhibition. Data indicate that BRCA1/2 deficient cancers express higher levels of neoantigens, thereby making themselves more immunogenic. The DNA damage created by PARP inhibitors generates an interferon response that leads to increased T-cell recruitment and TILs [113]. For example, preclinical studies demonstrate synergy between PARP inhibition and anti-CTLA-4 therapy in BRCA1/2 mutant ovarian cancer [114]. An interaction between PARP inhibitor and tumor-associated immunosuppression likely provides evidence to support the combination of PARP inhibitors and anti-PD-1/PD-L1 combinations. PARP inhibitor-related upregulation of PD-L1 expression in breast cancer cell lines and animal models appears to occur by knocking out GSK3β activity, which significantly increases PD-L1 expression and resistance to PARP inhibition. Hence, the blockade of PD-L1 re-sensitized tumor cells to PARP inhibition [115]. In an ongoing Phase II study at our institution, we are studying the combination of pembrolizumab and olaparib as subsequent-line therapy in BTC (NCT04306367) [116]. A separate Phase II study is looking at combined durvalumab plus olaparib in IDH-mutated solid tumors, including a cohort of patients with BTC (NCT03991832) [117].
Even more pertinent to the HRR pathway, several inhibitors of specific HRR genes, such as ATM, are now under preclinical development and early clinical investigation. A common practice in studying isolated genes in basic and translational research is to artificially knock out the gene of interest and observe downstream effects. What if we artificially induce DDR in tumor cells to increase their susceptibility to synthetic lethality with PARPi? Current ATM inhibitors include KU-60019 and AZD0156, which, when combined with PARPi in HRR stable cell lines, induce a massive increase in DNA damage and dysregulate the G2 DNA damage checkpoint [118]. This synergy and checkpoint dysregulation is also seen when a CHK1 inhibitor (PF-477736) was combined with rucaparib in cell lines [119].
12. Conclusions
Many factors limit the advancement of therapeutics in BTC, including anatomical challenges prohibiting ready access to tissue biopsy, locally advanced disease precluding curative resection, and the complicated biology of this aggressive cancer. Thus, there is an urgent need to develop new strategies to anticipate a BTC diagnosis at an early and resectable stage and obtain sufficient tissue samples to perform genomic analysis. Still, these limitations and needs should not distract from our recent achievements, including the incorporation of targeted agents and immunotherapies into our treatment arsenal and their use in place of traditional chemotherapy. Moreover, the mechanisms and intricacies of the HRR pathway continue to be elucidated. There is increasing evidence that the BRCA phenotype leading to HRD is not exclusive to BRCA1/2 mutations but rather encompasses many more genes in the HRR pathway. However, we recognize that each of the genes involved in HRR may not have equal significance, and there are varying degrees of preclinical and clinical evidence for the degree of contribution of each to BRCAness [120].
Our review is timely due to a better understanding of HRR, advancements in molecular profiling of cancers, and the availability of therapeutic agents targeting HRD cancer. Particularly in BTC, harboring one of the highest frequencies of HRR aberrations, the suboptimal standard chemotherapy represents an urgent call for newer approaches to cancer therapy. PARPi is the first class of drugs used clinically to target HRD cancers via synthetic lethality and has been well studied in BRCA1/2 mutated cancers, including pancreas, prostate, breast, and ovarian, generally with very positive results. In BTC, BRCA-mutant tumors occur at a frequency of <5%, but aberrations in HRR genes overall make up >25% of cases [22,23]. Although most data are currently limited to animal models and cell lines, we explored evidence supporting a role for several more frequently mutated HRR genes and their possible mechanisms of action in creating BRCAness in BTC. Ultimately, we encourage prospective studies that redefine and expand the qualification of patients for DDR-targeting drugs such as PARPi and investigate strategies to augment the effectiveness of these inhibitors.
Author Contributions
Conceptualization A.R.H. and G.M.K.; Methodology A.R.H., G.M.K. and C.Y.; Software B.D., S.A. and M.M.; formal analysis B.D., S.A., M.M. and R.C.J.; Resources B.D., S.A., M.M. and R.C.J.; Investigation C.Y., M.M. and T.R.; Writing-original draft preparation C.Y., M.K. and T.R.; Writing-review and editing A.R.H., G.M.K., C.Y., B.D., S.A. and R.C.J.; Visualization C.Y., G.M.K., A.R.H., M.M. and R.C.J.; Supervision A.R.H. and G.M.K. All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by the Ruesch Center for the Cure of Gastrointestinal Cancers and R01CA168635. The case included in the manuscript was treatment in an IRB-approved clinical trial sponsored by Merck (NCT04306367).
Institutional Review Board Statement
Not applicable. The authors de-identified the single patient who’s clinical course was described in this review article. No investigational intervention was performed on this patient.
Informed Consent Statement
Ethical review and approval were waived for this study due to de-identification of the single patient who’s clinical course was described in this review article. No investigational intervention was performed on the patient.
Data Availability Statement
This article is intented to be a review article; no investigational data was reported.
Acknowledgments
The authors would like to thank Marion L Hartley, PhD, for editing this manuscript.
Conflicts of Interest
The authors have no conflict of interest.
References
- Malaguarnera, G.; Giordano, M.; Paladina, I.; Rando, A.; Uccello, M.; Basile, F.; Biondi, A.; Carnazzo, S.; Alessandria, I.; Mazzarino, C. Markers of bile duct tumors. World J. Gastrointest. Oncol. 2011, 3, 49–59. [Google Scholar] [CrossRef]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Everhart, J.E.; Ruhl, C.E. Burden of Digestive Diseases in the United States Part III: Liver, Biliary Tract, and Pancreas. Gastroenterology 2009, 136, 1134–1144. [Google Scholar] [CrossRef]
- Forner, A.; Vidili, G.; Rengo, M.; Bujanda, L.; Ponz-Sarvisé, M.; Lamarca, A. Clinical presentation, diagnosis and staging of cholangiocarcinoma. Liver Int. 2019, 39, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Koerkamp, B.G.; Wiggers, J.K.; Gönen, M.; Doussot, A.; Allen, P.J.; Besselink, M.G.H.; Blumgart, L.H.; Busch, O.R.C.; D’Angelica, M.I.; DeMatteo, R.P.; et al. Survival after resection of perihilar cholangiocarcinoma—Development and external validation of a prognostic nomogram. Ann. Oncol. 2016, 27, 753. [Google Scholar] [CrossRef] [Green Version]
- DeOliveira, M.L.; Cunningham, S.C.; Cameron, J.L.; Kamangar, F.; Winter, J.M.; Lillemoe, K.D.; Choti, M.A.; Yeo, C.J.; Schulick, R.D. Cholangiocarcinoma. Ann. Surg. 2007, 245, 755–762. [Google Scholar] [CrossRef]
- Lubienski, A. Hepatocellular Carcinoma: Interventional Bridging to Liver Transplantation. Transplantation 2005, 80, S113–S119. [Google Scholar] [CrossRef]
- Endo, I.; Gonen, M.; Yopp, A.C.; Dalal, K.M.; Zhou, Q.; Klimstra, D.; D’Angelica, M.; DeMatteo, R.P.; Fong, Y.; Schwartz, L.; et al. Intrahepatic Cholangiocarcinoma. Ann. Surg. 2008, 248, 84–96. [Google Scholar] [CrossRef]
- Spolverato, G.; Kim, Y.; Alexandrescu, S.; Marques, H.P.; Lamelas, J.; Aldrighetti, L.; Gamblin, T.C.; Maithel, S.K.; Pulitano, C.; Bauer, T.W.; et al. Management and Outcomes of Patients with Recurrent Intrahepatic Cholangiocarcinoma Following Previous Curative-Intent Surgical Resection. Ann. Surg. Oncol. 2015, 23, 235–243. [Google Scholar] [CrossRef]
- Jarnagin, W.R.; Ruo, L.; Little, S.A.; Klimstra, D.; D’Angelica, M.; DeMatteo, R.P.; Wagman, R.; Blumgart, L.H.; Fong, Y. Patterns of initial disease recurrence after resection of gallbladder carcinoma and hilar cholangiocarcinoma: Implications for adjuvant therapeutic strategies. Cancer 2003, 98, 1689–1700. [Google Scholar] [CrossRef]
- Hasegawa, S.; Ikai, I.; Fujii, H.; Hatano, E.; Shimahara, Y. Surgical Resection of Hilar Cholangiocarcinoma: Analysis of Survival and Postoperative Complications. World J. Surg. 2007, 31, 1258–1265. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus Gemcitabine versus Gemcitabine for Biliary Tract Cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): A Phase 3, Open-;Label, Randomised, Controlled Trial. Lancet Oncol. 2021, 22, 690–701. [Google Scholar] [CrossRef]
- Park, I.; Lee, J.-L.; Ryu, M.-H.; Kim, T.-W.; Lee, S.S.; Park, D.H.; Lee, S.S.; Seo, D.W.; Kim, M.-H. Prognostic factors and predictive model in patients with advanced biliary tract adenocarcinoma receiving first-line palliative chemotherapy. Cancer 2009, 115, 4148–4155. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Ni Huang, M.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [Green Version]
- Farshidfar, F.; Zheng, S.; Gingras, M.-C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 19, 2878–2880. [Google Scholar] [CrossRef]
- Fiste, O.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.; Zagouri, F. The Emerging Role of Immunotherapy in Intrahepatic Cholangiocarcinoma. Vaccines 2021, 9, 422. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Brandi, G. Pemigatinib: Hot topics behind the first approval of a targeted therapy in cholangiocarcinoma. Cancer Treat. Res. Commun. 2021, 27, 100337. [Google Scholar] [CrossRef]
- Zhu, A.X.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.T.; Borad, M.J.; Bridgewater, J.A.; et al. Final Overall Survival Efficacy Results of Ivosidenib for Patients with Advanced Cholangiocarcinoma With IDH1 Mutation. JAMA Oncol. 2021, 7, 1669. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A Multicentre, Open-Label, Phase 2 Study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- COSMIC: The Catalogue of Somatic Mutations in Cancer. Available online: Cancer.sanger.ac.uk (accessed on 25 March 2022).
- Yang, W.; Sun, Y. Promising Molecular Targets for the Targeted Therapy of Biliary Tract Cancers: An Overview. OncoTargets Ther. 2021, 14, 1341–1366. [Google Scholar] [CrossRef]
- Ricci, A.D.; Rizzo, A.; Bonucci, C.; Tober, N.; Palloni, A.; Mollica, V.; Maggio, I.; Deserti, M.; Tavolari, S.; Brandi, G. PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block? Medicines 2020, 7, 54. [Google Scholar] [CrossRef]
- Minten, E.V.; Yu, D.S. DNA Repair: Translation to the Clinic. Clin. Oncol. 2019, 31, 303–310. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Goldstein, M.; Kastan, M.B. The DNA Damage Response: Implications for Tumor Responses to Radiation and Chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Caja, F.; Vodickova, L.; Kral, J.; Vymetalkova, V.; Naccarati, A.; Vodicka, P. DNA Mismatch Repair Gene Variants in Sporadic Solid Cancers. Int. J. Mol. Sci. 2020, 21, 5561. [Google Scholar] [CrossRef]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.-M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [Green Version]
- Mullen, J.; Kato, S.; Sicklick, J.K.; Kurzrock, R. Targeting ARID1A mutations in cancer. Cancer Treat. Rev. 2021, 100, 102287. [Google Scholar] [CrossRef]
- Park, Y.; Chui, M.H.; Rahmanto, Y.S.; Yu, Z.-C.; Shamanna, R.A.; Bellani, M.A.; Gaillard, S.; Ayhan, A.; Viswanathan, A.; Seidman, M.M.; et al. Loss of ARID1A in Tumor Cells Renders Selective Vulnerability to Combined Ionizing Radiation and PARP Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5584–5594. [Google Scholar] [CrossRef]
- Hu, H.-M.; Zhao, X.; Kaushik, S.; Robillard, L.; Barthelet, A.; Lin, K.K.; Shah, K.N.; Simmons, A.D.; Raponi, M.; Harding, T.C.; et al. A Quantitative Chemotherapy Genetic Interaction Map Reveals Factors Associated with PARP Inhibitor Resistance. Cell Rep. 2018, 23, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. The Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, Y.W.A.; Casals, T.; Carmichael, J.; Rodrigues, D.N.; et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Gout, J.; Perkhofer, L.; Morawe, M.; Arnold, F.; Ihle, M.; Biber, S.; Lange, S.; Roger, E.; Kraus, J.M.; Stifter, K.; et al. Synergistic targeting and resistance to PARP inhibition in DNA damage repair-deficient pancreatic cancer. Gut 2021, 70, 743–760. [Google Scholar] [CrossRef]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Xu, R.-H.; Chin, K.; Lee, K.-W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.-A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs kinases—The lessons from the mouse models: Inhibition ≠ deletion. Cell Biosci. 2020, 10, 81. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Juhász, S.; Elbakry, A.; Mathes, A.; Löbrich, M. ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol. Cell 2018, 71, 11–24.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbarino, J.; Eckroate, J.; Sundaram, R.K.; Jensen, R.B.; Bindra, R.S. Loss of ATRX confers DNA repair defects and PARP inhibitor sensitivity. Transl. Oncol. 2021, 14, 101147. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, I.H.; Davidson, R.; Gagné, J.-P.; Xu, Z.Z.; Poirier, G.G.; Hendzel, M.J. Germline Mutations in BAP1 Impair Its Function in DNA Double-Strand Break Repair. Cancer Res. 2014, 74, 4282–4294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbatino, F.; Liguori, L.; Malapelle, U.; Schiavi, F.; Tortora, V.; Conti, V.; Filippelli, A.; Tortora, G.; Ferrone, C.R.; Pepe, S. Case Report: BAP1 Mutation and RAD21 Amplification as Predictive Biomarkers to PARP Inhibitor in Metastatic Intrahepatic Cholangiocarcinoma. Front. Oncol. 2020, 10, 567289. [Google Scholar] [CrossRef]
- A Fennell, D.; King, A.; Mohammed, S.; Branson, A.; Brookes, C.; Darlison, L.; Dawson, A.G.; Gaba, A.; Hutka, M.; Morgan, B.; et al. Rucaparib in patients with BAP1-deficient or BRCA1-deficient mesothelioma (MiST1): An Open-Label, Single-Arm, Phase 2a Clinical Trial. Lancet Respir. Med. 2021, 9, 593–600. [Google Scholar] [CrossRef]
- Fabbro, M.; Rodriguez, J.A.; Baer, R.; Henderson, B.R. BARD1 Induces BRCA1 Intranuclear Foci Formation by Increasing RING-dependent BRCA1 Nuclear Import and Inhibiting BRCA1 Nuclear Export. J. Biol. Chem. 2002, 277, 21315–21324. [Google Scholar] [CrossRef] [Green Version]
- Özden, O.; Bishehsari, F.; Bauer, J.; Park, S.-H.; Jana, A.; Baik, S.H.; Sporn, J.C.; Staudacher, J.; Yazici, C.; Krett, N.; et al. Expression of an Oncogenic BARD1 Splice Variant Impairs Homologous Recombination and Predicts Response to PARP-1 Inhibitor Therapy in Colon Cancer. Sci. Rep. 2016, 6, 26273. [Google Scholar] [CrossRef]
- Kong, E.A.Y.; Xu, C.; Sun, X.; Sun, H.; Zhao, X.; He, N.; Ji, K.; Wang, Q.; Du, L.; Wang, J.; et al. BLM helicase inhibition synergizes with PARP inhibition to improve the radiosensitivity of olaparib resistant non-small cell lung cancer cells by inhibiting homologous recombination repair. Cancer Biol. Med. 2021, 18, 1–32. [Google Scholar] [CrossRef]
- Wang, C.-X.; Zhang, Z.-L.; Yin, Q.-K.; Tu, J.-L.; Wang, J.-E.; Xu, Y.-H.; Rao, Y.; Ou, T.-M.; Huang, S.-L.; Li, D.; et al. Design, Synthesis, and Evaluation of New Quinazolinone Derivatives that Inhibit Bloom Syndrome Protein (BLM) Helicase, Trigger DNA Damage at the Telomere Region, and Synergize with PARP Inhibitors. J. Med. Chem. 2020, 63, 9752–9772. [Google Scholar] [CrossRef]
- Smith, J.; Mun Tho, L.; Xu, N.A.; Gillespie, D. Chapter 3—The ATM–Chk2 and ATR–Chk1 pathways in DNA damage signaling and cancer. In Advances in Cancer Research; Vande Woude, G.F., Klein, G., Eds.; Academic Press: Cambridge, MA, USA, 2010; pp. 73–112. [Google Scholar]
- Yin, Y.; Shen, Q.; Zhang, P.; Tao, R.; Chang, W.; Li, R.; Xie, G.; Liu, W.; Zhang, L.; Kapoor, P.; et al. Chk1 inhibition potentiates the therapeutic efficacy of PARP inhibitor BMN673 in gastric cancer. Am. J. Cancer Res. 2017, 7, 473–483. [Google Scholar] [PubMed]
- Walden, H.; Deans, A.J. The Fanconi Anemia DNA Repair Pathway: Structural and Functional Insights into a Complex Disorder. Annu. Rev. Biophys. 2014, 43, 257–278. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women with Ovarian Cancer. JNCI J. Natl. Cancer Inst. 2015, 107, djv214. [Google Scholar] [CrossRef]
- Lajud, S.A.; Nagda, D.A.; Yamashita, T.; Zheng, J.; Tanaka, N.; Abuzeid, W.M.; Civantos, A.; Bezpalko, O.; O’Malley, B.W.; Li, D. Dual Disruption of DNA Repair and Telomere Maintenance for the Treatment of Head and Neck Cancer. Clin. Cancer Res. 2014, 20, 6465–6478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef]
- Alblihy, A.; Alabdullah, M.L.; Toss, M.S.; Algethami, M.; Mongan, N.P.; Rakha, E.A.; Madhusudan, S. RAD50 deficiency is a predictor of platinum sensitivity in sporadic epithelial ovarian cancers. Mol. Biomed. 2020, 1, 19. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, G.; Xue, F.; Edwards, R.; Sood, A.K.; Zhang, W.; Yang, D. Copy number deletion of RAD50 as predictive marker of BRCAness and PARP inhibitor response in BRCA wild type ovarian cancer. Gynecol. Oncol. 2016, 141, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Vispe, S.; Cazaux, C.; Lesca, C.; Defais, M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res. 1998, 26, 2859–2864. [Google Scholar] [CrossRef] [Green Version]
- Baumann, P.; West, S.C. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci. 1998, 23, 247–251. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.; Oliver, A.G.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.; Cohen, O.; Kochupurakkal, B.; Kim, D.; Dunn, C.; Buendia, J.B.; Wander, S.; Helvie, K.; Lloyd, M.; Marini, L.; et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol. 2020, 31, 590–598. [Google Scholar] [CrossRef]
- Gottifredi, V.; Wiesmüller, L. Current Understanding of RAD52 Functions: Fundamental and Therapeutic Insights. Cancers 2020, 12, 705. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.P. The Rad52–Rad59 complex interacts with Rad51 and replication protein A. DNA Repair 2003, 2, 1127–1134. [Google Scholar] [CrossRef]
- Toma, M.; Sullivan-Reed, K.; Śliwiński, T.; Skorski, T. RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers 2019, 11, 1561. [Google Scholar] [CrossRef] [Green Version]
- Sullivan-Reed, K.; Bolton-Gillespie, E.; Dasgupta, Y.; Langer, S.; Siciliano, M.; Nieborowska-Skorska, M.; Hanamshet, K.; Belyaeva, E.A.; Bernhardy, A.J.; Lee, J.; et al. Simultaneous Targeting of PARP1 and RAD52 Triggers Dual Synthetic Lethality in BRCA-Deficient Tumor Cells. Cell Rep. 2018, 23, 3127–3136. [Google Scholar] [CrossRef]
- Shamanna, R.A.; Lu, H.; de Freitas, J.K.; Tian, J.; Croteau, D.L.; Bohr, V.A. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat. Commun. 2016, 7, 13785. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Sinha, D.; Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Asaithamby, A. Werner Syndrome Protein and DNA Replication. Int. J. Mol. Sci. 2018, 19, 3442. [Google Scholar] [CrossRef] [Green Version]
- Arai, A.; Chano, T.; Futami, K.; Furuichi, Y.; Ikebuchi, K.; Inui, T.; Tameno, H.; Ochi, Y.; Shimada, T.; Hisa, Y.; et al. RECQL1 and WRN Proteins Are Potential Therapeutic Targets in Head and Neck Squamous Cell Carcinoma. Cancer Res. 2011, 71, 4598–4607. [Google Scholar] [CrossRef] [Green Version]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Camacho, C.V.; Kraus, W.L. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp. Mol. Med. 2021, 53, 42–51. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef] [Green Version]
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A., III; Bidziński, M.; Kim, J.-W.; Nam, J.H.; Madry, R.; Hernández, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients With Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Spizzo, G.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; Arora, S.P.; Khushman, M.M.; Salem, M.E.; Battaglin, F.; et al. Frequency of BRCA mutation in biliary tract cancer and its correlation with tumor mutational burden (TMB) and microsatellite instability (MSI). J. Clin. Oncol. 2019, 37, 4085. [Google Scholar] [CrossRef]
- Zhao, S.; Xu, Y.; Wu, W.; Wang, P.; Wang, Y.; Jiang, H.; Zhu, J. ARID1A Variations in Cholangiocarcinoma: Clinical Significances and Molecular Mechanisms. Front. Oncol. 2021, 11, 2360. [Google Scholar] [CrossRef]
- Sun, X.; Wang, S.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.-C.; et al. Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell 2017, 32, 574–589.e6. [Google Scholar] [CrossRef] [Green Version]
- Trizzino, M.; Barbieri, E.; Petracovici, A.; Wu, S.; Welsh, S.A.; Owens, T.A.; Licciulli, S.; Zhang, R.; Gardini, A. The Tumor Suppressor ARID1A Controls Global Transcription via Pausing of RNA Polymerase II. Cell Rep. 2018, 23, 3933–3945. [Google Scholar] [CrossRef]
- Reske, J.J.; Wilson, M.R.; Holladay, J.; Siwicki, R.A.; Skalski, H.; Harkins, S.; Adams, M.; Risinger, J.I.; Hostetter, G.; Lin, K.; et al. Co-existing TP53 and ARID1A mutations promote aggressive endometrial tumorigenesis. PLoS Genet. 2021, 17, e1009986. [Google Scholar] [CrossRef]
- Chaisaingmongkol, J.; Budhu, A.; Dang, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudhisawasdi, V.; Lertprasertsuke, N.; et al. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017, 32, 57–70.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, R. ARID1A loss in cancer: Towards a mechanistic understanding. Pharmacol. Ther. 2018, 190, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Zhao, T.; Tse, K.-H.; Chow, H.-M.; Cui, Y.; Jiang, L.; Du, S.; Loy, M.M.T.; Herrup, K. ATM and ATR play complementary roles in the behavior of excitatory and inhibitory vesicle populations. Proc. Natl. Acad. Sci. USA 2018, 115, E292–E301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, Y.-J.; Im, S.-A.; Lee, K.-W.; Cho, J.Y.; Song, E.-K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, Double-Blind Phase II Trial with Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients with Recurrent or Metastatic Gastric Cancer. J. Clin. Oncol. 2015, 33, 3858–3865. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ye, D.; Guan, K.-L.; Xiong, Y. IDH1 and IDH2 Mutations in Tumorigenesis: Mechanistic Insights and Clinical Perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Li, W.Y.; Tseng, A.; Beerman, I.; Elia, A.J.; Bendall, S.C.; Lemonnier, F.; Kron, K.J.; Cescon, D.W.; Hao, Z.; et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET. Cancer Cell 2016, 30, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Eder, J.P.; Doroshow, D.B.; Do, K.T.; Keedy, V.L.; Sklar, J.S.; Glazer, P.; Bindra, R.; Shapiro, G.I. Clinical Efficacy of Olaparib in IDH1/IDH2-Mutant Mesenchymal Sarcomas. JCO Precis. Oncol. 2021, 5, 466–472. [Google Scholar] [CrossRef]
- Zan, H.; Tat, C.; Qiu, Z.; Taylor, J.R.; Guerrero, J.A.; Shen, T.; Casali, P. Rad52 competes with Ku70/Ku86 for binding to S-region DSB ends to modulate antibody class-switch DNA recombination. Nat. Commun. 2017, 8, 14244. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.E.; Meghani, K.; Brault, M.-E.; Leclerc, L.; He, Y.; Day, T.A.; Elias, K.M.; Drapkin, R.; Weinstock, D.M.; Dao, F.; et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell Rep. 2016, 14, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Murfuni, I.; Rass, U. Chapter 8—Targeting homologous recombination repair in cancer. In DNA Repair in Cancer Therapy, 2nd ed.; Kelley, M.R., Fishel, M.L., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 225–275. [Google Scholar]
- Graeser, M.; McCarthy, A.; Lord, C.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.S.; et al. A Marker of Homologous Recombination Predicts Pathologic Complete Response to Neoadjuvant Chemotherapy in Primary Breast Cancer. Clin. Cancer Res. 2010, 16, 6159–6168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tennstedt, P.; Fresow, R.; Simon, R.; Marx, A.; Terracciano, L.; Petersen, C.; Sauter, G.; Dikomey, E.; Borgmann, K. RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int. J. Cancer 2012, 132, 2118–2126. [Google Scholar] [CrossRef] [PubMed]
- Wiegmans, A.P.; Al-Ejeh, F.; Chee, N.; Yap, P.-Y.; Gorski, J.J.; Da Silva, L.; Bolderson, E.; Chenevix-Trench, G.; Anderson, R.; Simpson, P.T.; et al. Rad51 supports triple negative breast cancer metastasis. Oncotarget 2014, 5, 3261–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.-L.; Liu, S.-S.; Xiong, Z.-F.; Wang, F.; Li, X.-Y.; Deng, H. Clinical significance of RAD51C and its contribution to ovarian carcinogenesis. Int. J. Clin. Exp. Pathol. 2020, 13, 14–20. [Google Scholar] [PubMed]
- Suwaki, N.; Klare, K.; Tarsounas, M. RAD51 paralogs: Roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 2011, 22, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Rein, H.L.; A Bernstein, K.; A Baldock, R. RAD51 paralog function in replicative DNA damage and tolerance. Curr. Opin. Genet. Dev. 2021, 71, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Song, H.; Leslie, G.; Engel, C.; Hahnen, E.; Auber, B.; Horváth, J.; Kast, K.; Niederacher, D.; Turnbull, C.; et al. Ovarian and Breast Cancer Risks Associated With Pathogenic Variants in RAD51C and RAD51D. JNCI J. Natl. Cancer Inst. 2020, 112, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; McInerny, S.; Zethoven, M.; Cheasley, D.; Lim, B.W.X.; Rowley, S.M.; Devereux, L.; Grewal, N.; Ahmadloo, S.; Byrne, D.; et al. Combined Tumor Sequencing and Case-Control Analyses of RAD51C in Breast Cancer. J. Natl. Cancer Inst. 2019, 111, 1332–1338. [Google Scholar] [CrossRef]
- Boussios, S.; Karathanasi, A.; Cooke, D.; Neille, C.; Sadauskaite, A.; Moschetta, M.; Zakynthinakis-Kyriakou, N.; Pavlidis, N. PARP Inhibitors in Ovarian Cancer: The Route to “Ithaca”. Diagnostics 2019, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Min, A.; Im, S.-A.; Yoon, Y.-K.; Song, S.-H.; Nam, H.-J.; Hur, H.-S.; Kim, H.-P.; Lee, K.-H.; Han, S.-W.; Oh, D.-Y.; et al. RAD51C-Deficient Cancer Cells Are Highly Sensitive to the PARP Inhibitor Olaparib. Mol. Cancer Ther. 2013, 12, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, E.S.; Pavlick, D.; Khiabanian, H.; Frampton, G.M.; Ross, J.S.; Gregg, J.P.; Lara, P.N.; Oesterreich, S.; Agarwal, N.; Necchi, A.; et al. Pan-Cancer Analysis of BRCA1 and BRCA2 Genomic Alterations and Their Association with Genomic Instability as Measured by Genome-Wide Loss of Heterozygosity. JCO Precis. Oncol. 2020, 4, 442–465. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An International, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Westphalen, C.B.; Fine, A.D.; André, F.; Ganesan, S.; Heinemann, V.; Rouleau, E.; Turnbull, C.; Palacios, L.G.; Lopez, J.-A.; Sokol, E.S.; et al. Pan-cancer Analysis of Homologous Recombination Repair–associated Gene Alterations and Genome-wide Loss-of-Heterozygosity Score. Clin. Cancer Res. 2021, 28, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary Somatic Mutations Restoring BRCA1/2 Predict Chemotherapy Resistance in Hereditary Ovarian Carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klotz, D.M.; Wimberger, P. Overcoming PARP inhibitor resistance in ovarian cancer: What Are the Most Promising Strategies? Arch. Gynecol. Obstet. 2020, 302, 1087–1102. [Google Scholar] [CrossRef]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [Green Version]
- A Konstantinopoulos, P.; Barry, W.T.; Birrer, M.; Westin, S.N.; A Cadoo, K.; I Shapiro, G.; Mayer, E.L.; E O’Cearbhaill, R.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A Dose-Escalation and Dose-Expansion Phase 1b Trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Mehta, R.; Wood, A.C.; Yu, J.; Kim, R. Investigational PARP inhibitors for the treatment of biliary tract cancer: Spotlight on Preclinical and Clinical Studies. Expert Opin. Investig. Drugs 2021, 30, 451–461. [Google Scholar] [CrossRef]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Armstrong, S.A.; Agarwal, S.; Wang, H.; Noel, M.S.; Weinberg, B.A.; Marshall, J.; He, A.R. Phase II study of combination pembrolizumab and olaparib in patients with advanced cholangiocarcinoma: Interim results. J. Clin. Oncol. 2022, 40, 452. [Google Scholar] [CrossRef]
- Chen, E.X.; O’Kane, G.M.; Mason, W.P.; Knox, J.J.; Razak, A.R.A.; Zadeh, G. Phase II basket trial of olaparib and durvalumab in patients (pts) with isocitrate dehydrogenase (IDH) mutated solid tumors. J. Clin. Oncol. 2020, 38, TPS3167. [Google Scholar] [CrossRef]
- Mak, J.P.; Ma, H.T.; Poon, R.Y. Synergism between ATM and PARP1 Inhibition Involves DNA Damage and Abrogating the G2 DNA Damage Checkpoint. Mol. Cancer Ther. 2019, 19, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.L.; Prendergast, L.; Curtin, N.J. Exploring the Synergy between PARP and CHK1 Inhibition in Matched BRCA2 Mutant and Corrected Cells. Cancers 2020, 12, 878. [Google Scholar] [CrossRef] [Green Version]
- Ricci, A.D.; Rizzo, A.; Brandi, G. The DNA damage repair (DDR) pathway in biliary tract cancer (BTC): A New Pandora’s Box? ESMO Open 2020, 5, e001042. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).