
Ring Finger Protein 125 Is an Anti-Proliferative Tumor Suppressor in Hepatocellular Carcinoma

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Lentiviral Production and Transduction
2.3. Xenograft Assay
2.4. siRNA Transfection
2.5. WST-1 Cell Proliferation Assay
2.6. RT-qPCR and RNA-Seq
2.7. Western Blot
2.8. Bioinformatic Analysis
2.9. Statistics
3. Results
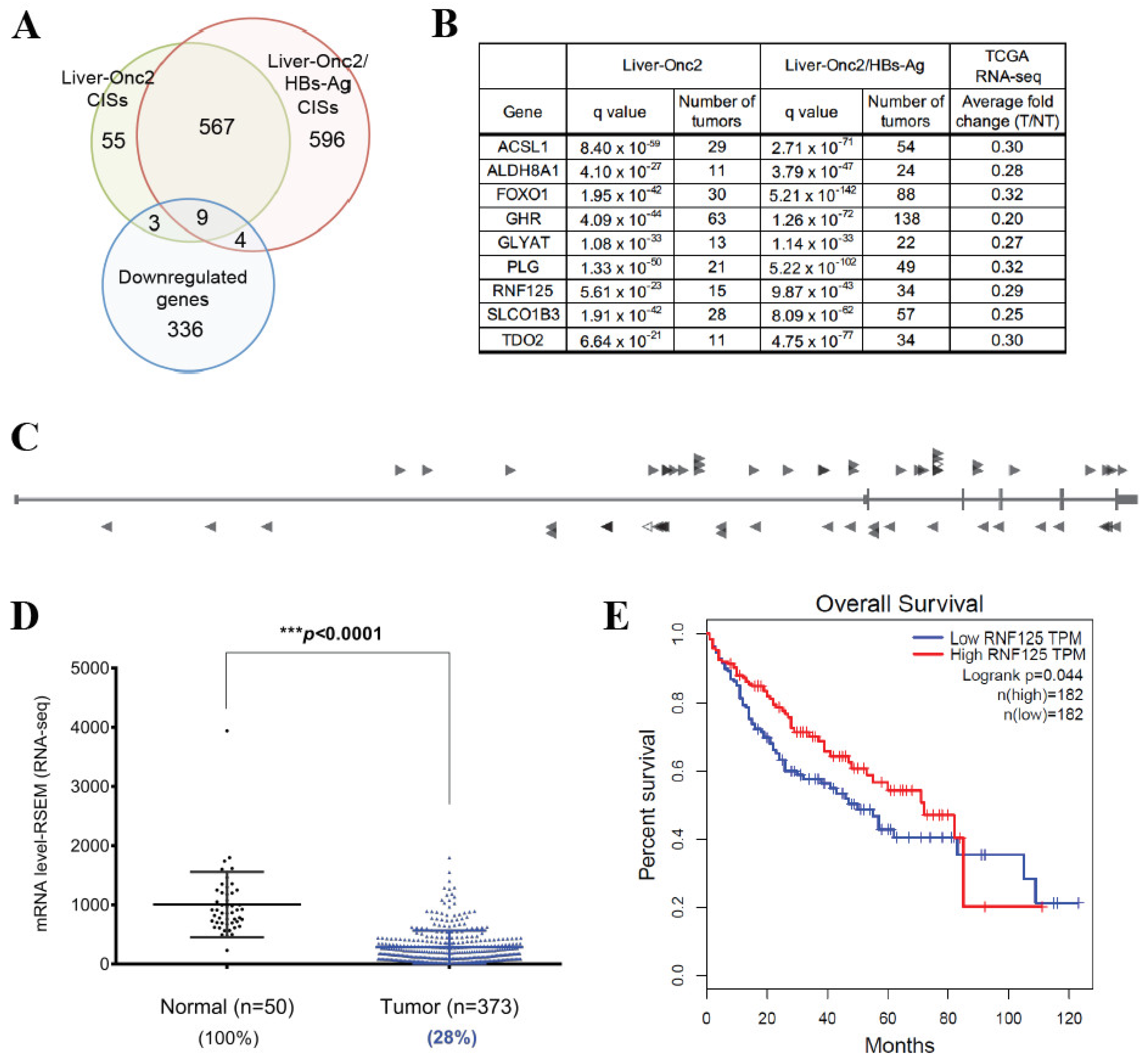
3.1. RNF125 Is a Candidate Tumor Suppressor and Significantly Downregulated in Human HCC
3.2. RNF125 Expression in Tumor Tissues Showed a Significant Negative Correlation with Survival in HCC Patients
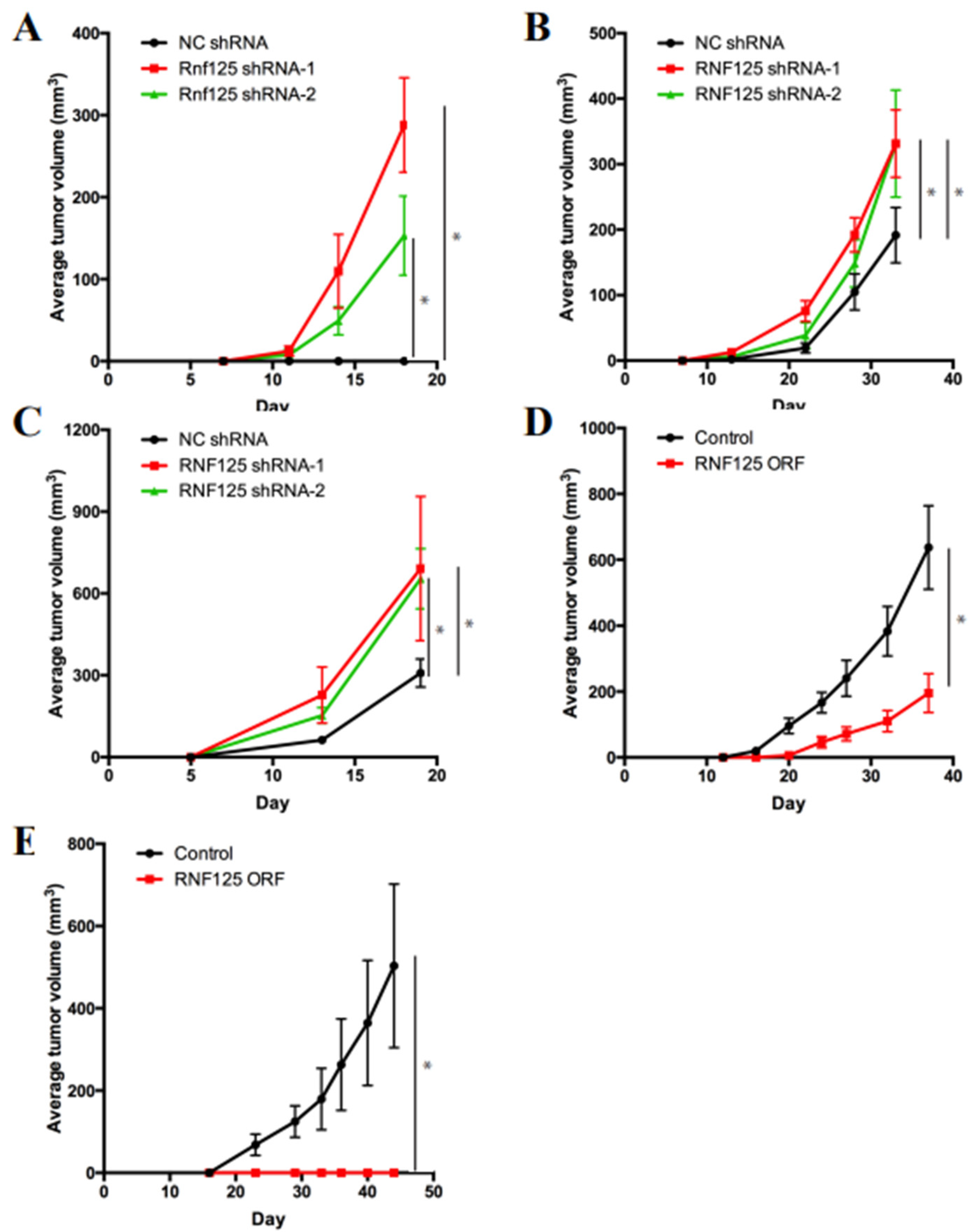
3.3. RNF125 Is a Bona Fide Tumor Suppressor in Mouse and Human HCC
3.4. RNF125 Negatively Regulates Cell Proliferation In Vitro
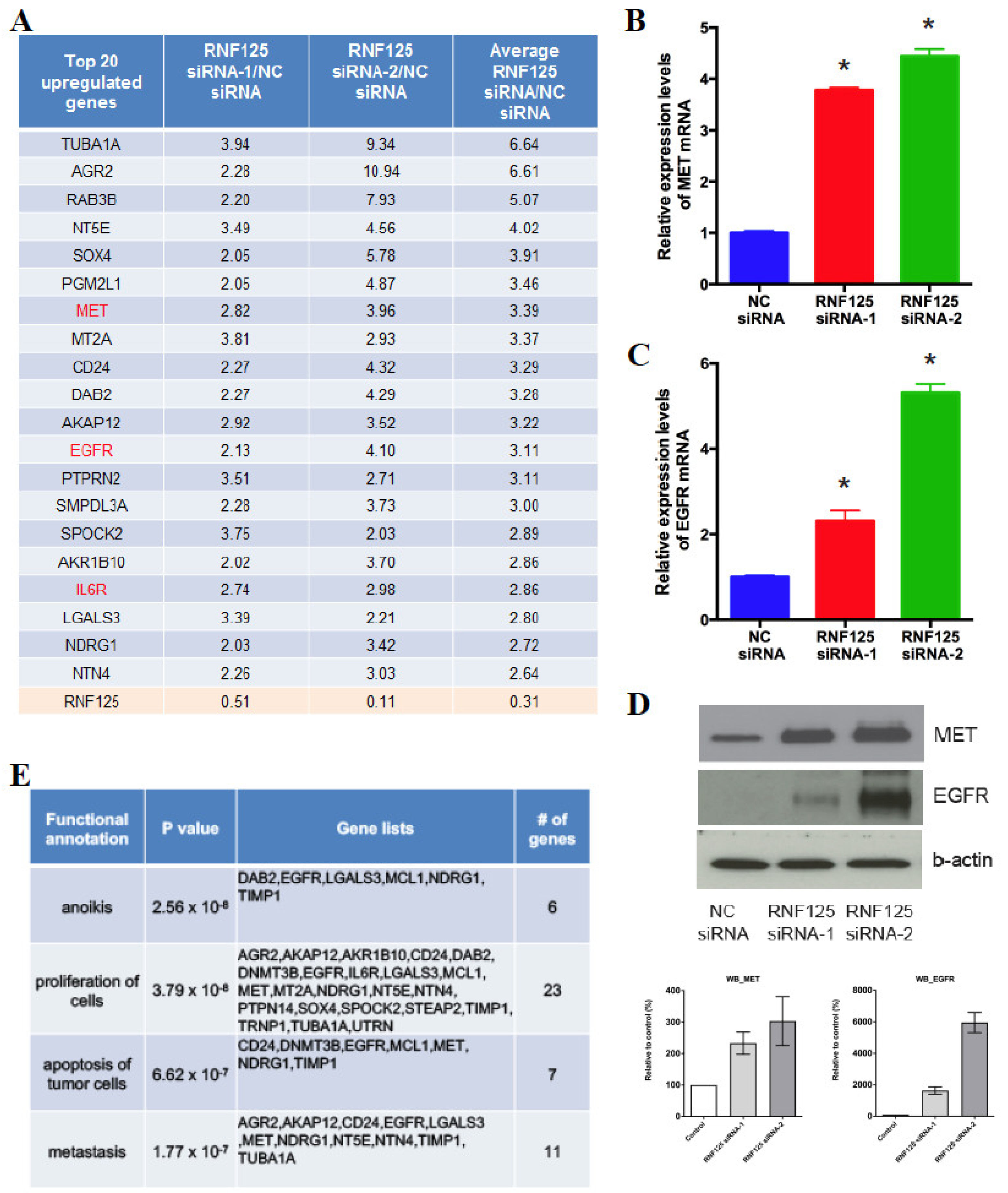
3.5. RNF125 Transcriptionally Suppresses the Global Cell Proliferation Machinery
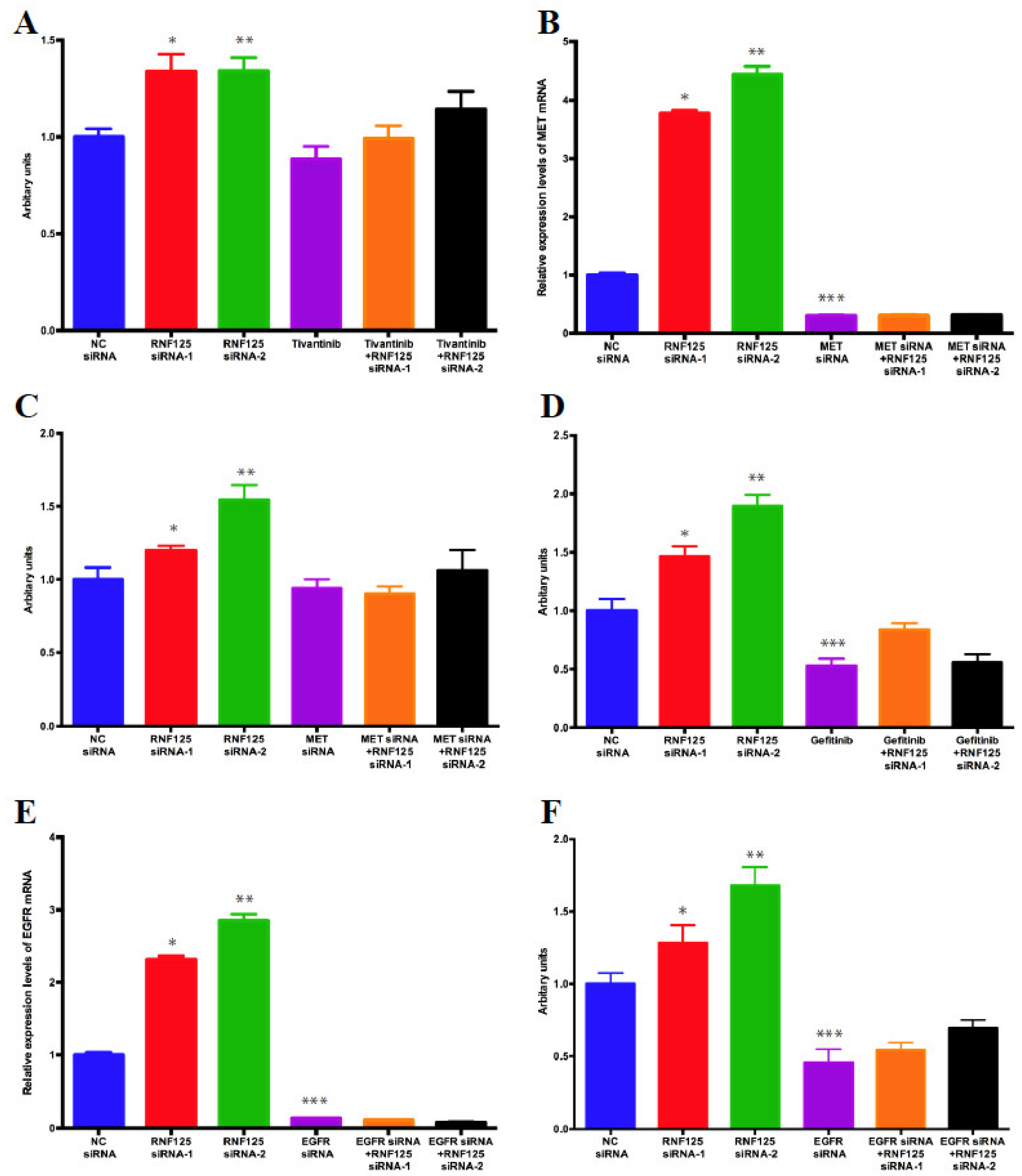
3.6. RNF125 Negatively Regulates Cell Proliferation through Inhibition of the MET and EGFR Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Henshaw, C.; Youlden, D.R.; Clark, P.J.; Aitken, J.F.; Baade, P.D. Global Trends in Incidence Rates of Primary Adult Liver Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; El-Serag, H.B. The Epidemiology of Hepatocellular Carcinoma in the USA. Curr. Gastroenterol. Rep. 2019, 21, 17. [Google Scholar] [CrossRef]
- International Cancer Genome, C.; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Shibata, T. Comprehensive genome sequencing of the liver cancer genome. Cancer Lett. 2013, 340, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Bard-Chapeau, E.A.; Nguyen, A.T.; Rust, A.G.; Sayadi, A.; Lee, P.; Chua, B.Q.; New, L.S.; de Jong, J.; Ward, J.M.; Chin, C.K.; et al. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. Nat. Genet. 2014, 46, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Zender, L.; Xue, W.; Zuber, J.; Semighini, C.P.; Krasnitz, A.; Ma, B.; Zender, P.; Kubicka, S.; Luk, J.M.; Schirmacher, P.; et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 2008, 135, 852–864. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucl. Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Chisari, F.V.; Klopchin, K.; Moriyama, T.; Pasquinelli, C.; Dunsford, H.A.; Sell, S.; Pinkert, C.A.; Brinster, R.L.; Palmiter, R.D. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 1989, 59, 1145–1156. [Google Scholar] [CrossRef]
- Collier, L.S.; Carlson, C.M.; Ravimohan, S.; Dupuy, A.J.; Largaespada, D.A. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature 2005, 436, 272–276. [Google Scholar] [CrossRef]
- Dupuy, A.J.; Akagi, K.; Largaespada, D.A.; Copeland, N.G.; Jenkins, N.A. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 2005, 436, 221–226. [Google Scholar] [CrossRef]
- Dupuy, A.J.; Rogers, L.M.; Kim, J.; Nannapaneni, K.; Starr, T.K.; Liu, P.; Largaespada, D.A.; Scheetz, T.E.; Jenkins, N.A.; Copeland, N.G. A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res. 2009, 69, 8150–8156. [Google Scholar] [CrossRef] [Green Version]
- Keng, V.W.; Villanueva, A.; Chiang, D.Y.; Dupuy, A.J.; Ryan, B.J.; Matise, I.; Silverstein, K.A.; Sarver, A.; Starr, T.K.; Akagi, K.; et al. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat. Biotechnol. 2009, 27, 264–274. [Google Scholar] [CrossRef]
- Mann, K.M.; Ward, J.M.; Yew, C.C.; Kovochich, A.; Dawson, D.W.; Black, M.A.; Brett, B.T.; Sheetz, T.E.; Dupuy, A.J.; Australian Pancreatic Cancer Genome Initiative; et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 5934–5941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, M.B.; Black, M.A.; Jones, D.J.; Ward, J.M.; Yew, C.C.; Newberg, J.Y.; Dupuy, A.J.; Rust, A.G.; Bosenberg, M.W.; McMahon, M.; et al. Transposon mutagenesis identifies genetic drivers of Braf(V600E) melanoma. Nat. Genet. 2015, 47, 486–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- March, H.N.; Rust, A.G.; Wright, N.A.; ten Hoeve, J.; de Ridder, J.; Eldridge, M.; van der Weyden, L.; Berns, A.; Gadiot, J.; Uren, A.; et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat. Genet. 2011, 43, 1202–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Mancera, P.A.; Rust, A.G.; van der Weyden, L.; Kristiansen, G.; Li, A.; Sarver, A.L.; Silverstein, K.A.; Grutzmann, R.; Aust, D.; Rummele, P.; et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 2012, 486, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Rad, R.; Rad, L.; Wang, W.; Cadinanos, J.; Vassiliou, G.; Rice, S.; Campos, L.S.; Yusa, K.; Banerjee, R.; Li, M.A.; et al. PiggyBac transposon mutagenesis: A tool for cancer gene discovery in mice. Science 2010, 330, 1104–1107. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Wei, Z.; Koso, H.; Rust, A.G.; Yew, C.C.; Mann, M.B.; Ward, J.M.; Adams, D.J.; Copeland, N.G.; Jenkins, N.A. Transposon mutagenesis identifies genes and evolutionary forces driving gastrointestinal tract tumor progression. Nat. Genet. 2015, 47, 142–150. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lee, G.R.; Woo, D.H.; Park, N.H.; Cha, H.J.; Moon, Y.H.; Han, I.S. Depletion of Aurora A leads to upregulation of FoxO1 to induce cell cycle arrest in hepatocellular carcinoma cells. Cell Cycle 2013, 12, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, J.H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.W.; Shen, G.Z.; Cao, L.Q.; Jiang, X.F.; Peng, H.P.; Shen, G.; Chen, D.; Xue, P. MicroRNA-1269 promotes proliferation in human hepatocellular carcinoma via downregulation of FOXO1. BMC Cancer 2014, 14, 909. [Google Scholar] [CrossRef] [Green Version]
- Koso, H.; Takeda, H.; Yew, C.C.; Ward, J.M.; Nariai, N.; Ueno, K.; Nagasaki, M.; Watanabe, S.; Rust, A.G.; Adams, D.J.; et al. Transposon mutagenesis identifies genes that transform neural stem cells into glioma-initiating cells. Proc. Natl. Acad. Sci. USA 2012, 109, E2998–E3007. [Google Scholar] [CrossRef] [Green Version]
- Koso, H.; Tsuhako, A.; Lyons, E.; Ward, J.M.; Rust, A.G.; Adams, D.J.; Jenkins, N.A.; Copeland, N.G.; Watanabe, S. Identification of FoxR2 as an oncogene in medulloblastoma. Cancer Res. 2014, 74, 2351–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Unno, M.; Onogawa, T.; Tokui, T.; Kondo, T.N.; Nakagomi, R.; Adachi, H.; Fujiwara, K.; Okabe, M.; Suzuki, T.; et al. LST-2, a human liver-specific organic anion transporter, determines methotrexate sensitivity in gastrointestinal cancers. Gastroenterology 2001, 120, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Barbosa, E.; Singh, I. Molecular cloning and sequencing of human palmitoyl-CoA ligase and its tissue specific expression. Mol. Cell. Biochem. 1995, 151, 77–81. [Google Scholar] [CrossRef]
- Lin, M.; Napoli, J.L. cDNA cloning and expression of a human aldehyde dehydrogenase (ALDH) active with 9-cis-retinal and identification of a rat ortholog, ALDH12. J. Biol. Chem. 2000, 275, 40106–40112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mawal, Y.R.; Qureshi, I.A. Purification to homogeneity of mitochondrial acyl coa:glycine n-acyltransferase from human liver. Biochem. Biophys. Res. Commun. 1994, 205, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Rafice, S.A.; Chauhan, N.; Efimov, I.; Basran, J.; Raven, E.L. Oxidation of L-tryptophan in biology: A comparison between tryptophan 2,3-dioxygenase and indoleamine 2,3-dioxygenase. Biochem. Soc. Trans. 2009, 37, 408–412. [Google Scholar] [CrossRef] [Green Version]
- Mueller, K.M.; Kornfeld, J.W.; Friedbichler, K.; Blaas, L.; Egger, G.; Esterbauer, H.; Hasselblatt, P.; Schlederer, M.; Haindl, S.; Wagner, K.U.; et al. Impairment of hepatic growth hormone and glucocorticoid receptor signaling causes steatosis and hepatocellular carcinoma in mice. Hepatology 2011, 54, 1398–1409. [Google Scholar] [CrossRef]
- Lin, C.C.; Liu, T.W.; Yeh, M.L.; Tsai, Y.S.; Tsai, P.C.; Huang, C.F.; Huang, J.F.; Chuang, W.L.; Dai, C.Y.; Yu, M.L. Significant down-regulation of growth hormone receptor expression revealed as a new unfavorable prognostic factor in hepatitis C virus-related hepatocellular carcinoma. Clin. Mol. Hepatol. 2021, 27, 313–328. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Tenorio, J.; Mansilla, A.; Valencia, M.; Martinez-Glez, V.; Romanelli, V.; Arias, P.; Castrejon, N.; Poletta, F.; Guillen-Navarro, E.; Gordo, G.; et al. A new overgrowth syndrome is due to mutations in RNF125. Hum. Mutat. 2014, 35, 1436–1441. [Google Scholar] [CrossRef]
- Arimoto, K.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannini, A.L.; Gao, Y.; Bijlmakers, M.J. T-cell regulator RNF125/TRAC-1 belongs to a novel family of ubiquitin ligases with zinc fingers and a ubiquitin-binding domain. Biochem. J. 2008, 410, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver regeneration. Hepatology 2006, 43, S45–S53. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.; Pardo, J.; Zhao, H.; Li, C.C.; Pali, E.; Shen, M.M.; Qu, K.; Yu, S.X.; Huang, B.C.; Yu, P.; et al. Systematic identification of regulatory proteins critical for T-cell activation. J. Biol. 2003, 2, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Li, C.C.; Pardo, J.; Chu, P.C.; Liao, C.X.; Huang, J.; Dong, J.G.; Zhou, X.; Huang, Q.; Huang, B.; et al. A novel E3 ubiquitin ligase TRAC-1 positively regulates T cell activation. J. Immunol. 2005, 174, 5288–5297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoji-Kawata, S.; Zhong, Q.; Kameoka, M.; Iwabu, Y.; Sapsutthipas, S.; Luftig, R.B.; Ikuta, K. The RING finger ubiquitin ligase RNF125/TRAC-1 down-modulates HIV-1 replication in primary human peripheral blood mononuclear cells. Virology 2007, 368, 191–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Tu, S.; Lin, G.; Guo, H.; Yan, C.; Liu, Q.; Huang, L.; Tang, N.; Xiao, Y.; Pope, R.M.; et al. Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.; Zhou, H.; Wu, C.; Wu, Q.; Ma, S.; Wei, C.; Cao, Y.; Song, J.; Zhong, H.; Zhou, Z.; et al. The Ubiquitin Ligase RNF125 Targets Innate Immune Adaptor Protein TRIM14 for Ubiquitination and Degradation. J. Immunol. 2017, 198, 4652–4658. [Google Scholar] [CrossRef]
- Kim, H.; Frederick, D.T.; Levesque, M.P.; Cooper, Z.A.; Feng, Y.; Krepler, C.; Brill, L.; Samuels, Y.; Hayward, N.K.; Perlina, A.; et al. Downregulation of the Ubiquitin Ligase RNF125 Underlies Resistance of Melanoma Cells to BRAF Inhibitors via JAK1 Deregulation. Cell Rep. 2015, 11, 1458–1473. [Google Scholar] [CrossRef] [Green Version]
- Zender, L.; Villanueva, A.; Tovar, V.; Sia, D.; Chiang, D.Y.; Llovet, J.M. Cancer gene discovery in hepatocellular carcinoma. J. Hepatol. 2010, 52, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Puri, N.; Salgia, R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J. Carcinog. 2008, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Stolz, D.B.; Esplen, J.E.; Dorko, K.; Michalopoulos, G.K.; Strom, S.C. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J. Biol. Chem. 2000, 275, 8806–8811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Guan, G.; Zhang, J.; Zheng, H.; Li, D.; Wang, W.; Lu, F.; Chen, X. E2F1-mediated AUF1 upregulation promotes HCC development and enhances drug resistance via stabilization of AKR1B10. Cancer Sci. 2022, 113, 1154–1167. [Google Scholar] [CrossRef] [PubMed]
- Hikita, H.; Takehara, T.; Shimizu, S.; Kodama, T.; Shigekawa, M.; Iwase, K.; Hosui, A.; Miyagi, T.; Tatsumi, T.; Ishida, H.; et al. The Bcl-xL inhibitor, ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib. Hepatology 2010, 52, 1310–1321. [Google Scholar] [CrossRef]
- Hao, F.; Fei, X.; Ren, X.; Xi Xiao, J.; Chen, Y.; Wang, J. Pseudogene AKR1B10P1 enhances tumorigenicity and regulates epithelial-mesenchymal transition in hepatocellular carcinoma via stabilizing SOX4. J. Cell. Mol. Med. 2020, 24, 11779–11790. [Google Scholar] [CrossRef]
- Xie, X.; Rigor, P.; Baldi, P. MotifMap: A human genome-wide map of candidate regulatory motif sites. Bioinformatics 2009, 25, 167–174. [Google Scholar] [CrossRef]
- Essaghir, A.; Toffalini, F.; Knoops, L.; Kallin, A.; van Helden, J.; Demoulin, J.B. Transcription factor regulation can be accurately predicted from the presence of target gene signatures in microarray gene expression data. Nucl. Acids Res. 2010, 38, e120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Xu, L.; Sun, C. Comprehensive characterization of cancer genes in hepatocellular carcinoma genomes. Oncol. Lett. 2018, 15, 1503–1510. [Google Scholar] [CrossRef]
- Villanueva, A.; Portela, A.; Sayols, S.; Battiston, C.; Hoshida, Y.; Méndez-González, J.; Imbeaud, S.; Letouzé, E.; Hernandez-Gea, V.; Cornella, H.; et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 2015, 61, 1945–1956. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Ye, J.; Nie, Y.; Ashraf, U.; Zohaib, A.; Duan, X.; Fu, Z.F.; Song, Y.; Chen, H.; Cao, S. MicroRNA-15b Modulates Japanese Encephalitis Virus-Mediated Inflammation via Targeting RNF125. J. Immunol. 2015, 195, 2251–2262. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kodama, T.; Kodama, M.; Jenkins, N.A.; Copeland, N.G.; Chen, H.J.; Wei, Z. Ring Finger Protein 125 Is an Anti-Proliferative Tumor Suppressor in Hepatocellular Carcinoma. Cancers 2022, 14, 2589. https://doi.org/10.3390/cancers14112589
Kodama T, Kodama M, Jenkins NA, Copeland NG, Chen HJ, Wei Z. Ring Finger Protein 125 Is an Anti-Proliferative Tumor Suppressor in Hepatocellular Carcinoma. Cancers. 2022; 14(11):2589. https://doi.org/10.3390/cancers14112589
Chicago/Turabian StyleKodama, Takahiro, Michiko Kodama, Nancy A. Jenkins, Neal G. Copeland, Huanhuan Joyce Chen, and Zhubo Wei. 2022. "Ring Finger Protein 125 Is an Anti-Proliferative Tumor Suppressor in Hepatocellular Carcinoma" Cancers 14, no. 11: 2589. https://doi.org/10.3390/cancers14112589