
Tumor Necrosis Factor Receptor 2 (TNFR2): An Emerging Target in Cancer Therapy


Abstract
:Simple Summary
Abstract
1. Introduction
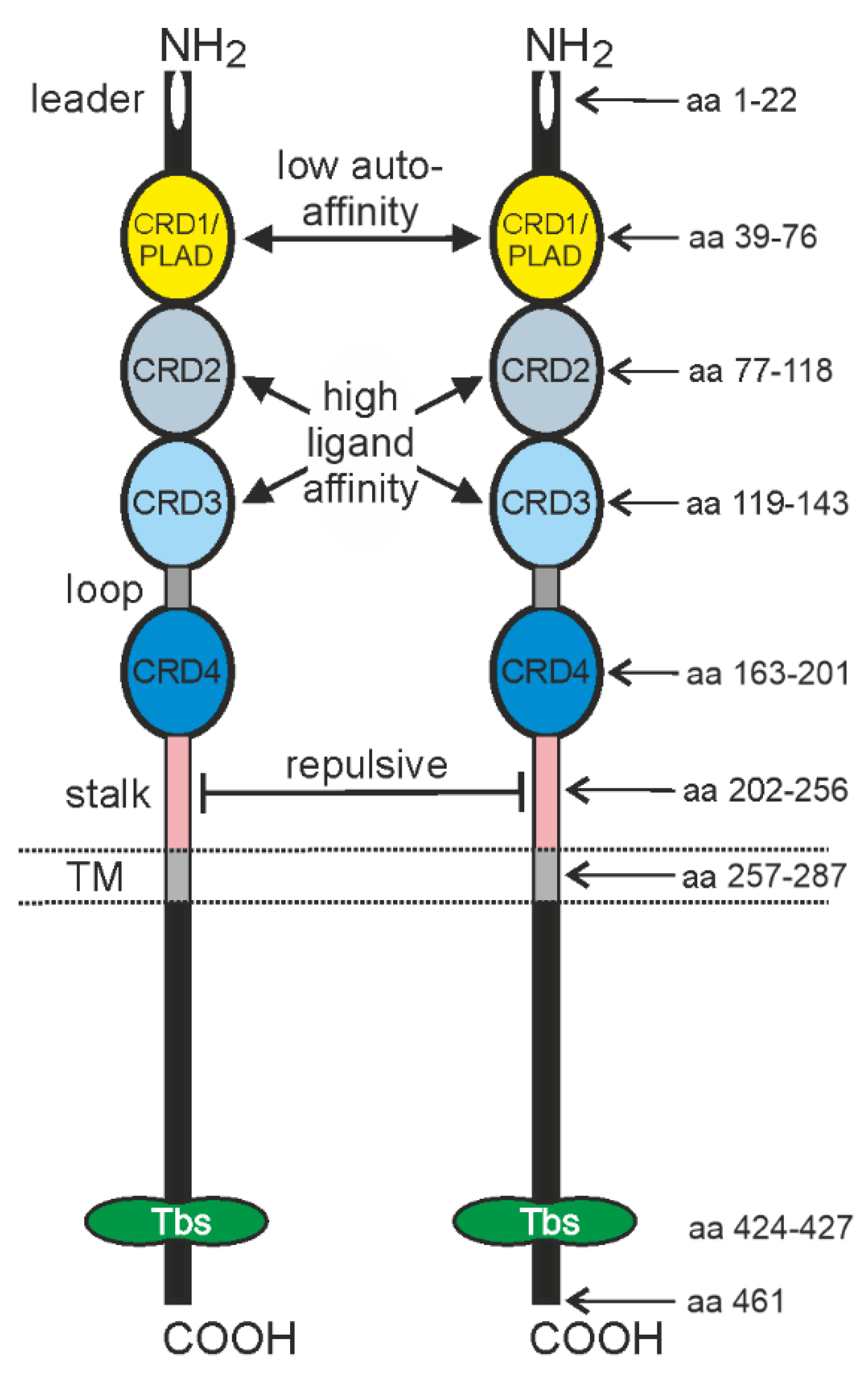
2. TNFR2 Signaling
3. TNFR2 in Immune Regulation
4. TNFR2 in Cancer
4.1. TNFR2 in Tumor Immune Escape
4.2. TNFR2 as An Oncogene
5. TNFR2 as a Therapeutic Target in Cancer
5.1. Antagonistic Anti-TNFR2 Antibodies in Preclinical Tumor Models
5.2. Anti-TNFR2 Antibody Agonism in Preclinical Tumor Models
5.3. TNFR2 Targeting and Immune Checkpoint Blockade
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Medler, J.; Wajant, H. Tumor necrosis factor receptor-2 (TNFR2): An overview of an emerging drug target. Expert Opin. Ther. Targets 2019, 23, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Nakamura, T.; Yoshikawa, M.; Yoshioka, Y.; Tsunoda, S.; Nakagawa, S.; Yamagata, Y.; Tsutsumi, Y. Solution of the structure of the TNF-TNFR2 complex. Sci. Signal. 2010, 3, ra83. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Messerschmidt, S.; Holeiter, G.; Tepperink, J.; Osswald, S.; Zappe, A.; Branschädel, M.; Boschert, V.; Mann, D.A.; Scheurich, P.; et al. The tumor necrosis factor receptor stalk regions define responsiveness to soluble versus membrane-bound ligand. Mol. Cell. Biol. 2012, 32, 2515–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucka, K.; Lang, I.; Zhang, T.; Siegmund, D.; Medler, J.; Wajant, H. Membrane lymphotoxin-α(2)β is a novel tumor necrosis factor (TNF) receptor 2 (TNFR2) agonist. Cell Death Dis. 2021, 12, 360. [Google Scholar] [CrossRef]
- Tang, P.; Hung, M.C.; Klostergaard, J. Human pro-tumor necrosis factor is a homotrimer. Biochemistry 1996, 35, 8216–8225. [Google Scholar] [CrossRef]
- Siegmund, D.; Kums, J.; Ehrenschwender, M.; Wajant, H. Activation of TNFR2 sensitizes macrophages for TNFR1-mediated necroptosis. Cell Death Dis. 2016, 7, e2375. [Google Scholar] [CrossRef]
- Wajant, H. The role of TNF in cancer. Results Probl. Cell Differ. 2009, 49, 1–15. [Google Scholar] [CrossRef]
- Moss, M.L.; Jin, S.L.; Milla, M.E.; Bickett, D.M.; Burkhart, W.; Carter, H.L.; Chen, W.J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Grell, M.; Douni, E.; Wajant, H.; Löhden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Mauri, D.N.; Ebner, R.; Montgomery, R.I.; Kochel, K.D.; Cheung, T.C.; Yu, G.L.; Ruben, S.; Murphy, M.; Eisenberg, R.J.; Cohen, G.H.; et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 1998, 8, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Wajant, H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015, 22, 1727–1741. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Meng, F.; Gao, X.; Dong, H.; Yao, W. Expression and purification of a natural N-terminal pre-ligand assembly domain of tumor necrosis factor receptor 1 (TNFR1 PLAD) and preliminary activity determination. Protein J. 2011, 30, 281–289. [Google Scholar] [CrossRef]
- Karathanasis, C.; Medler, J.; Fricke, F.; Smith, S.; Malkusch, S.; Widera, D.; Fulda, S.; Wajant, H.; van Wijk, S.J.L.; Dikic, I.; et al. Single-molecule imaging reveals the oligomeric state of functional TNFα-induced plasma membrane TNFR1 clusters in cells. Sci. Signal. 2020, 13, eaax5647. [Google Scholar] [CrossRef]
- Prada, J.P.; Wangorsch, G.; Kucka, K.; Lang, I.; Dandekar, T.; Wajant, H. A systems-biology model of the tumor necrosis factor (TNF) interactions with TNF receptor 1 and 2. Bioinformatics 2021, 37, 69–676. [Google Scholar] [CrossRef]
- Pan, S.; An, P.; Zhang, R.; He, X.; Yin, G.; Min, W. Etk/Bmx as a tumor necrosis factor receptor type 2-specific kinase: Role in endothelial cell migration and angiogenesis. Mol. Cell. Biol. 2002, 22, 7512–7523. [Google Scholar] [CrossRef] [Green Version]
- So, T.; Croft, M. Regulation of PI-3-Kinase and Akt Signaling in T Lymphocytes and Other Cells by TNFR Family Molecules. Front. Immunol. 2013, 4, 139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Xu, Y.; Ekman, N.; Wu, Z.; Wu, J.; Alitalo, K.; Min, W. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J. Biol. Chem. 2003, 278, 51267–51276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothe, M.; Wong, S.C.; Henzel, W.J.; Goeddel, D.V. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell 1994, 78, 681–692. [Google Scholar] [CrossRef]
- Borghi, A.; Haegman, M.; Fischer, R.; Carpentier, I.; Bertrand, M.J.M.; Libert, C.; Afonina, I.S.; Beyaert, R. The E3 ubiquitin ligases HOIP and cIAP1 are recruited to the TNFR2 signaling complex and mediate TNFR2-induced canonical NF-κB signaling. Biochem. Pharmacol. 2018, 153, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.J.; Conze, D.B.; Li, X.; Ying, S.X.; Hanover, J.A.; Ashwell, J.D. TNF-alpha induced c-IAP1/TRAF2 complex translocation to a Ubc6-containing compartment and TRAF2 ubiquitination. EMBO J. 2005, 24, 1886–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, Y.; Takahashi, M.; Morishita, T.; Noguchi., T.; Matsuzawa, A. Post-Translational Modifications of the TAK1-TAB Complex. Int. J. Mol. Sci. 2017, 18, 205. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef]
- Baldwin, A.S., Jr. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.H.; Keats, J.J.; Wang, H.; Vignali, D.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fotin-Mleczek, M.; Henkler, F.; Samel, D.; Reichwein, M.; Hausser, A.; Parmryd, I.; Scheurich, P.; Schmid, J.A.; Wajant, H. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J. Cell Sci. 2002, 115, 2757–2770. [Google Scholar] [CrossRef] [PubMed]
- Rauert, H.; Stühmer, T.; Bargou, R.; Wajant, H.; Siegmund, D. TNFR1 and TNFR2 regulate the extrinsic apoptotic pathway in myeloma cells by multiple mechanisms. Cell Death Dis. 2011, 2, e194. [Google Scholar] [CrossRef] [Green Version]
- Dueber, E.C.; Schoeffler, A.J.; Lingel, A.; Elliott, J.M.; Fedorova, A.V.; Giannetti, A.M.; Zobel, K.; Maurer, B.; Varfolomeev, E.; Wu, P.; et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science 2011, 334, 376–380. [Google Scholar] [CrossRef]
- Feltham, R.; Bettjeman, B.; Budhidarmo, R.; Mace, P.D.; Shirley, S.; Condon, S.M.; Chunduru, S.K.; McKinlay, M.A.; Vaux, D.L.; Silke, J.; et al. Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J. Biol. Chem. 2011, 286, 17015–17028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.; Kabaleeswaran, V.; Wang, Y.; Cheng, G.; Wu, H. Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: Affinity, specificity, and regulation. Mol. Cell 2010, 38, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Chau, D.; Callus, B.; Wong, W.W.; Hawkins, C.J.; Schneider, P.; McKinlay, M.; Benetatos, C.A.; Condon, S.M.; Chunduru, S.K.; et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J. Cell Biol. 2008, 182, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Duckett, C.S.; Thompson, C.B. CD30-dependent degradation of TRAF2: Implications for negative regulation of TRAF signaling and the control of cell survival. Genes Dev. 1997, 11, 2810–2821. [Google Scholar] [CrossRef] [Green Version]
- Grell, M.; Zimmermann, G.; Gottfried, E.; Chen, C.M.; Grunwald, U.; Huang, D.C.; Wu Lee, Y.H.; Durkop, H.; Engelmann, H.; Scheurich, P.; et al. Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: A role for TNF-R1 activation by endogenous membrane-anchored TNF. Embo J. 1999, 18, 3034–3043. [Google Scholar] [CrossRef] [Green Version]
- Wicovsky, A.; Henkler, F.; Salzmann, S.; Scheurich, P.; Kneitz, C.; Wajant, H. Tumor necrosis factor receptor-associated factor-1 enhances proinflammatory TNF receptor-2 signaling and modifies TNFR1-TNFR2 cooperation. Oncogene 2009, 28, 1769–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, T.; Grell, M.; Hessabi, B.; Bourteele, S.; Muller, G.; Scheurich, P.; Wajant, H. Enhancement of TNF receptor p60-mediated cytotoxicity by TNF receptor p80: Requirement of the TNF receptor-associated factor-2 binding site. J. Immunol. 1997, 158, 2398–2404. [Google Scholar] [PubMed]
- Schneider, P.; Schwenzer, R.; Haas, E.; Mühlenbeck, F.; Schubert, G.; Scheurich, P.; Tschopp, J.; Wajant, H. TWEAK can induce cell death via endogenous TNF and TNF receptor 1. Eur. J. Immunol. 1999, 29, 1785–1792. [Google Scholar] [CrossRef]
- Knop, J.; Spilgies, L.M.; Rufli, S.; Reinhart, R.; Vasilikos, L.; Yabal, M.; Owsley, E.; Jost, P.J.; Marsh, R.A.; Wajant, H.; et al. TNFR2 induced priming of the inflammasome leads to a RIPK1-dependent cell death in the absence of XIAP. Cell Death Dis. 2019, 10, 700. [Google Scholar] [CrossRef] [Green Version]
- Siegmund, D.; Ehrenschwender, M.; Wajant, H. TNFR2 unlocks a RIPK1 kinase activity-dependent mode of proinflammatory TNFR1 signaling. Cell Death Dis. 2018, 9, 921. [Google Scholar] [CrossRef]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Lazzeri, E.; Manetti, R.; Vanini, V.; Romagnani, P.; Maggi, E.; Romagnani, S. Phenotype, localization, and mechanism of suppression of CD4(+)CD25(+) human thymocytes. J. Exp. Med. 2002, 196, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Subleski, J.J.; Kopf, H.; Howard, O.M.; Mannel, D.N.; Oppenheim, J.J. Cutting edge: Expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: Applicability to tumor-infiltrating T regulatory cells. J. Immunol. 2008, 180, 6467–6471. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Bäumel, M.; Männel, D.N.; Howard, O.M.; Oppenheim, J.J. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol. 2007, 179, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef]
- Chen, X.; Hamano, R.; Subleski, J.J.; Hurwitz, A.A.; Howard, O.M.; Oppenheim, J.J. Expression of costimulatory TNFR2 induces resistance of CD4+FoxP3- conventional T cells to suppression by CD4+FoxP3+ regulatory T cells. J. Immunol. 2010, 185, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajant, H.; Beilhack, A. Targeting Regulatory T Cells by Addressing Tumor Necrosis Factor and Its Receptors in Allogeneic Hematopoietic Cell Transplantation and Cancer. Front. Immunol. 2019, 10, 2040. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Park, S.G.; Strickland, I.; Hayden, M.S.; Ghosh, S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 2009, 31, 921–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-κB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108.e1013. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.; Grinberg-Bleyer, Y.; Liao, W.; Maloney, D.; Wang, P.; Wu, Z.; Wang, J.; Bhatt, D.M.; Heise, N.; Schmid, R.M.; et al. An NF-κB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 2017, 47, 450–465.e455. [Google Scholar] [CrossRef] [Green Version]
- Lubrano di Ricco, M.; Ronin, E.; Collares, D.; Divoux, J.; Grégoire, S.; Wajant, H.; Gomes, T.; Grinberg-Bleyer, Y.; Baud, V.; Marodon, G.; et al. Tumor necrosis factor receptor family costimulation increases regulatory T-cell activation and function via NF-κB. Eur. J. Immunol. 2020, 50, 972–985. [Google Scholar] [CrossRef] [Green Version]
- Grinberg-Bleyer, Y.; Caron, R.; Seeley, J.J.; De Silva, N.S.; Schindler, C.W.; Hayden, M.S.; Klein, U.; Ghosh, S. The Alternative NF-κB Pathway in Regulatory T Cell Homeostasis and Suppressive Function. J. Immunol. 2018, 200, 2362–2371. [Google Scholar] [CrossRef]
- Tseng, W.Y.; Huang, Y.S.; Clanchy, F.; McNamee, K.; Perocheau, D.; Ogbechi, J.; Luo, S.F.; Feldmann, M.; McCann, F.E.; Williams, R.O. TNF receptor 2 signaling prevents DNA methylation at the Foxp3 promoter and prevents pathogenic conversion of regulatory T cells. Proc. Natl. Acad. Sci. USA 2019, 116, 21666–21672. [Google Scholar] [CrossRef]
- Urbano, P.C.M.; Koenen, H.; Joosten, I.; He, X. An Autocrine TNFα-Tumor Necrosis Factor Receptor 2 Loop Promotes Epigenetic Effects Inducing Human Treg Stability In Vitro. Front. Immunol. 2018, 9, 573. [Google Scholar] [CrossRef] [Green Version]
- Arvey, A.; van der Veeken, J.; Samstein, R.M.; Feng, Y.; Stamatoyannopoulos, J.A.; Rudensky, A.Y. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat. Immunol. 2014, 15, 580–587. [Google Scholar] [CrossRef] [Green Version]
- DuPage, M.; Chopra, G.; Quiros, J.; Rosenthal, W.L.; Morar, M.M.; Holohan, D.; Zhang, R.; Turka, L.; Marson, A.; Bluestone, J.A. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity 2015, 42, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Xiao, Y.; Hu, H.; Zou, Q.; Li, Y.; Gao, Y.; Ge, W.; Cheng, X.; Sun, S.C. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat. Commun. 2015, 6, 5930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Kivit, S.; Mensink, M.; Hoekstra, A.T.; Berlin, I.; Derks, R.J.E.; Both, D.; Aslam, M.A.; Amsen, D.; Berkers, C.R.; Borst, J. Stable human regulatory T cells switch to glycolysis following TNF receptor 2 costimulation. Nat. Metab. 2020, 2, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Ablamunits, V.; Bisikirska, B.; Herold, K.C. Acquisition of regulatory function by human CD8(+) T cells treated with anti-CD3 antibody requires TNF. Eur. J. Immunol. 2010, 40, 2891–2901. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, D.A.; Pan, S.; Ou, J.N.; Wang, J.; Chen, M.; Gray, J.D.; Zheng, S.G. Therapeutic polyclonal human CD8+ CD25+ Fox3+ TNFR2+ PD-L1+ regulatory cells induced ex-vivo. Clin. Immunol. 2013, 149, 450–463. [Google Scholar] [CrossRef] [Green Version]
- Aravena, O.; Pesce, B.; Soto, L.; Orrego, N.; Sabugo, F.; Wurmann, P.; Molina, M.C.; Alfaro, J.; Cuchacovich, M.; Aguillón, J.C.; et al. Anti-TNF therapy in patients with rheumatoid arthritis decreases Th1 and Th17 cell populations and expands IFN-γ-producing NK cell and regulatory T cell subsets. Immunobiology 2011, 216, 1256–1263. [Google Scholar] [CrossRef]
- Stoop, J.N.; Woltman, A.M.; Biesta, P.J.; Kusters, J.G.; Kuipers, E.J.; Janssen, H.L.; van der Molen, R.G. Tumor necrosis factor alpha inhibits the suppressive effect of regulatory T cells on the hepatitis B virus-specific immune response. Hepatology 2007, 46, 699–705. [Google Scholar] [CrossRef]
- Kim, E.Y.; Priatel, J.J.; Teh, S.J.; Teh, H.S. TNF receptor type 2 (p75) functions as a costimulator for antigen-driven T cell responses in vivo. J. Immunol. 2006, 176, 1026–1035. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Teh, H.S. TNF type 2 receptor (p75) lowers the threshold of T cell activation. J. Immunol. 2001, 167, 6812–6820. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.L.; Wei, X.S.; Zhang, M.; Niu, Y.R.; Zhou, Q. The Significance of Tumor Necrosis Factor Receptor Type II in CD8(+) Regulatory T Cells and CD8(+) Effector T Cells. Front. Immunol. 2018, 9, 583. [Google Scholar] [CrossRef] [Green Version]
- Afonina, I.S.; Cullen, S.P.; Martin, S.J. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol. Rev. 2010, 235, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Lee-Chang, C.; Bodogai, M.; Moritoh, K.; Chen, X.; Wersto, R.; Sen, R.; Young, H.A.; Croft, M.; Ferrucci, L.; Biragyn, A. Aging Converts Innate B1a Cells into Potent CD8+ T Cell Inducers. J. Immunol. 2016, 196, 3385–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Chang, C.; Bodogai, M.; Moritoh, K.; Olkhanud, P.B.; Chan, A.C.; Croft, M.; Mattison, J.A.; Holst, P.J.; Gress, R.E.; Ferrucci, L.; et al. Accumulation of 4-1BBL+ B cells in the elderly induces the generation of granzyme-B+ CD8+ T cells with potential antitumor activity. Blood 2014, 124, 1450–1459. [Google Scholar] [CrossRef]
- Kim, E.Y.; Teh, S.J.; Yang, J.; Chow, M.T.; Teh, H.S. TNFR2-deficient memory CD8 T cells provide superior protection against tumor cell growth. J. Immunol. 2009, 183, 6051–6057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teh, H.S.; Seebaran, A.; Teh, S.J. TNF receptor 2-deficient CD8 T cells are resistant to Fas/Fas ligand-induced cell death. J. Immunol. 2000, 165, 4814–4821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otano, I.; Alvarez, M.; Minute, L.; Ochoa, M.C.; Migueliz, I.; Molina, C.; Azpilikueta, A.; de Andrea, C.E.; Etxeberria, I.; Sanmamed, M.F.; et al. Human CD8 T cells are susceptible to TNF-mediated activation-induced cell death. Theranostics 2020, 10, 4481–4489. [Google Scholar] [CrossRef] [PubMed]
- Joedicke, J.J.; Myers, L.; Carmody, A.B.; Messer, R.J.; Wajant, H.; Lang, K.S.; Lang, P.A.; Mak, T.W.; Hasenkrug, K.J.; Dittmer, U. Activated CD8+ T cells induce expansion of Vβ5+ regulatory T cells via TNFR2 signaling. J. Immunol. 2014, 193, 2952–2960. [Google Scholar] [CrossRef] [Green Version]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Zhao, X.; Rong, L.; Zhao, X.; Li, X.; Liu, X.; Deng, J.; Wu, H.; Xu, X.; Erben, U.; Wu, P.; et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Investig. 2012, 122, 4094–4104. [Google Scholar] [CrossRef]
- Polz, J.; Remke, A.; Weber, S.; Schmidt, D.; Weber-Steffens, D.; Pietryga-Krieger, A.; Müller, N.; Ritter, U.; Mostböck, S.; Männel, D.N. Myeloid suppressor cells require membrane TNFR2 expression for suppressive activity. Immun. Inflamm. Dis. 2014, 2, 121–130. [Google Scholar] [CrossRef]
- Hu, X.; Li, B.; Li, X.; Zhao, X.; Wan, L.; Lin, G.; Yu, M.; Wang, J.; Jiang, X.; Feng, W.; et al. Transmembrane TNF-α promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J. Immunol. 2014, 192, 1320–1331. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Galan, L.; Vesin, D.; Uysal, H.; Blaser, G.; Benkhoucha, M.; Ryffel, B.; Quesniaux, V.F.J.; Garcia, I. Transmembrane Tumor Necrosis Factor Controls Myeloid-Derived Suppressor Cell Activity via TNF Receptor 2 and Protects from Excessive Inflammation during BCG-Induced Pleurisy. Front. Immunol. 2017, 8, 999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naserian, S.; Abdelgawad, M.E.; Afshar Bakshloo, M.; Ha, G.; Arouche, N.; Cohen, J.L.; Salomon, B.L.; Uzan, G. The TNF/TNFR2 signaling pathway is a key regulatory factor in endothelial progenitor cell immunosuppressive effect. Cell Commun. Signal. CCS 2020, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Hurrell, B.P.; Galle-Treger, L.; Jahani, P.S.; Howard, E.; Helou, D.G.; Banie, H.; Soroosh, P.; Akbari, O. TNFR2 Signaling Enhances ILC2 Survival, Function, and Induction of Airway Hyperreactivity. Cell Rep. 2019, 29, 4509–4524.e4505. [Google Scholar] [CrossRef] [Green Version]
- Razazian, M.; Khosravi, M.; Bahiraii, S.; Uzan, G.; Shamdani, S.; Naserian, S. Differences and similarities between mesenchymal stem cell and endothelial progenitor cell immunoregulatory properties against T cells. World J. Stem Cells 2021, 13, 971–984. [Google Scholar] [CrossRef]
- Beldi, G.; Khosravi, M.; Abdelgawad, M.E.; Salomon, B.L.; Uzan, G.; Haouas, H.; Naserian, S. TNFalpha/TNFR2 signaling pathway: An active immune checkpoint for mesenchymal stem cell immunoregulatory function. Stem Cell Res. Ther. 2020, 11, 281. [Google Scholar] [CrossRef] [PubMed]
- Beldi, G.; Bahiraii, S.; Lezin, C.; Nouri Barkestani, M.; Abdelgawad, M.E.; Uzan, G.; Naserian, S. TNFR2 Is a Crucial Hub Controlling Mesenchymal Stem Cell Biological and Functional Properties. Front. Cell Dev. Biol. 2020, 8, 596831. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Tran, L.; Torrey, H.; Song, Y.; Perkins, H.; Case, K.; Zheng, H.; Takahashi, H.; Kuhtreiber, W.M.; Faustman, D.L. Optimizing TNFR2 antagonism for immunotherapy with tumor microenvironment specificity. J. Leukoc. Biol. 2020, 107, 971–980. [Google Scholar] [CrossRef]
- Ticha, O.; Moos, L.; Wajant, H.; Bekeredjian-Ding, I. Expression of Tumor Necrosis Factor Receptor 2 Characterizes TLR9-Driven Formation of Interleukin-10-Producing B Cells. Front. Immunol. 2017, 8, 1951. [Google Scholar] [CrossRef]
- Schioppa, T.; Moore, R.; Thompson, R.G.; Rosser, E.C.; Kulbe, H.; Nedospasov, S.; Mauri, C.; Coussens, L.M.; Balkwill, F.R. B regulatory cells and the tumor-promoting actions of TNF-α during squamous carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 10662–10667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, M.W.; Ritchie, D.S.; Neeson, P.; Smyth, M.J. Biology and clinical observations of regulatory T cells in cancer immunology. Curr. Top. Microbiol. Immunol. 2011, 344, 61–95. [Google Scholar] [CrossRef] [PubMed]
- Ghods, A.; Mehdipour, F.; Shariat, M.; Talei, A.R.; Ghaderi, A. Regulatory T cells express Tumor Necrosis Factor Receptor 2 with the highest intensity among CD4(+) T cells in the draining lymph nodes of breast cancer. Mol. Immunol. 2021, 137, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, C.; Tian, T.; Zhang, T.; Wang, R.; Han, F.; Zhong, C.; Hua, M.; Ma, D. Increased Regulatory T Cells in Peripheral Blood of Acute Myeloid Leukemia Patients Rely on Tumor Necrosis Factor (TNF)-α-TNF Receptor-2 Pathway. Front. Immunol. 2018, 9, 1274. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.L.; Peng, W.B.; Niu, Y.R.; Xiang, X.; Wei, X.S.; Wang, Z.H.; Wang, X.; Zhang, S.Y.; Chen, X.; Zhou, Q. Accumulation of TNFR2-expressing regulatory T cells in malignant pleural effusion of lung cancer patients is associated with poor prognosis. Ann. Transl. Med. 2020, 8, 1647. [Google Scholar] [CrossRef]
- Yan, F.; Du, R.; Wei, F.; Zhao, H.; Yu, J.; Wang, C.; Zhan, Z.; Ding, T.; Ren, X.; Chen, X.; et al. Expression of TNFR2 by regulatory T cells in peripheral blood is correlated with clinical pathology of lung cancer patients. Cancer Immunol. Immunother. CII 2015, 64, 1475–1485. [Google Scholar] [CrossRef]
- Baram, T.; Erlichman, N.; Dadiani, M.; Balint-Lahat, N.; Pavlovski, A.; Meshel, T.; Morzaev-Sulzbach, D.; Gal-Yam, E.N.; Barshack, I.; Ben-Baruch, A. Chemotherapy Shifts the Balance in Favor of CD8+ TNFR2+ TILs in Triple-Negative Breast Tumors. Cells 2021, 10, 1429. [Google Scholar] [CrossRef]
- Okubo, Y.; Mera, T.; Wang, L.; Faustman, D.L. Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci. Rep. 2013, 3, 3153. [Google Scholar] [CrossRef] [Green Version]
- Nomelini, R.S.; Borges Júnior, L.E.; de Lima, C.A.; Chiovato, A.F.C.; Micheli, D.C.; Tavares-Murta, B.M.; Murta, E.F.C. TNF-R2 in tumor microenvironment as prognostic factor in epithelial ovarian cancer. Clin. Exp. Med. 2018, 18, 547–554. [Google Scholar] [CrossRef]
- Govindaraj, C.; Scalzo-Inguanti, K.; Madondo, M.; Hallo, J.; Flanagan, K.; Quinn, M.; Plebanski, M. Impaired Th1 immunity in ovarian cancer patients is mediated by TNFR2+ Tregs within the tumor microenvironment. Clin. Immunol. 2013, 149, 97–110. [Google Scholar] [CrossRef]
- Torrey, H.; Butterworth, J.; Mera, T.; Okubo, Y.; Wang, L.; Baum, D.; Defusco, A.; Plager, S.; Warden, S.; Huang, D.; et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci. Signal. 2017, 10, eaaf8608. [Google Scholar] [CrossRef] [PubMed]
- Kampan, N.C.; Madondo, M.T.; McNally, O.M.; Stephens, A.N.; Quinn, M.A.; Plebanski, M. Interleukin 6 Present in Inflammatory Ascites from Advanced Epithelial Ovarian Cancer Patients Promotes Tumor Necrosis Factor Receptor 2-Expressing Regulatory T Cells. Front. Immunol. 2017, 8, 1482. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinante, F.; Rigo, A.; Tecchio, C.; Morosato, L.; Nadali, G.; Ricetti, M.M.; Krampera, M.; Zanolin, E.; Locatelli, F.; Gallati, H.; et al. Serum levels of p55 and p75 soluble TNF receptors in adult acute leukaemia at diagnosis: Correlation with clinical and biological features and outcome. Br. J. Haematol. 1998, 102, 1025–1034. [Google Scholar] [CrossRef] [Green Version]
- Volk, A.; Li, J.; Xin, J.; You, D.; Zhang, J.; Liu, X.; Xiao, Y.; Breslin, P.; Li, Z.; Wei, W.; et al. Co-inhibition of NF-κB and JNK is synergistic in TNF-expressing human AML. J. Exp. Med. 2014, 211, 1093–1108. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Volk, A.; Zhang, J.; Cannova, J.; Dai, S.; Hao, C.; Hu, C.; Sun, J.; Xu, Y.; Wei, W.; et al. Sensitizing leukemia stem cells to NF-κB inhibitor treatment in vivo by inactivation of both TNF and IL-1 signaling. Oncotarget 2017, 8, 8420–8435. [Google Scholar] [CrossRef] [Green Version]
- Kagoya, Y.; Yoshimi, A.; Kataoka, K.; Nakagawa, M.; Kumano, K.; Arai, S.; Kobayashi, H.; Saito, T.; Iwakura, Y.; Kurokawa, M. Positive feedback between NF-κB and TNF-α promotes leukemia-initiating cell capacity. J. Clin. Investig. 2014, 124, 528–542. [Google Scholar] [CrossRef]
- Xin, J.; You, D.; Breslin, P.; Li, J.; Zhang, J.; Wei, W.; Cannova, J.; Volk, A.; Gutierrez, R.; Xiao, Y.; et al. Sensitizing acute myeloid leukemia cells to induced differentiation by inhibiting the RIP1/RIP3 pathway. Leukemia 2017, 31, 1154–1165. [Google Scholar] [CrossRef] [Green Version]
- Höckendorf, U.; Yabal, M.; Herold, T.; Munkhbaatar, E.; Rott, S.; Jilg, S.; Kauschinger, J.; Magnani, G.; Reisinger, F.; Heuser, M.; et al. RIPK3 Restricts Myeloid Leukemogenesis by Promoting Cell Death and Differentiation of Leukemia Initiating Cells. Cancer Cell 2016, 30, 75–91. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Shao, Y.L.; Wang, L.L.; Lin, J.; Zhang, J.B.; Ding, Y.; Gao, B.B.; Liu, D.H.; Gao, X.N. YTHDF2 is a potential target of AML1/ETO-HIF1α loop-mediated cell proliferation in t(8;21) AML. Oncogene 2021, 40, 3786–3798. [Google Scholar] [CrossRef]
- Paris, J.; Morgan, M.; Campos, J.; Spencer, G.J.; Shmakova, A.; Ivanova, I.; Mapperley, C.; Lawson, H.; Wotherspoon, D.A.; Sepulveda, C.; et al. Targeting the RNA m(6)A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 2019, 25, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safferthal, C.; Rohde, K.; Fulda, S. Therapeutic targeting of necroptosis by Smac mimetic bypasses apoptosis resistance in acute myeloid leukemia cells. Oncogene 2017, 36, 1487–1502. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.H.; Herrera, A.H.; Li, Y.; Walcheck, B. Role of ADAM17 in the ectodomain shedding of TNF-alpha and its receptors by neutrophils and macrophages. J. Leukoc. Biol. 2007, 82, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Torrey, H.; Kühtreiber, W.M.; Okubo, Y.; Tran, L.; Case, K.; Zheng, H.; Vanamee, E.; Faustman, D.L. A novel TNFR2 agonist antibody expands highly potent regulatory T cells. Sci. Signal. 2020, 13, eaba9600. [Google Scholar] [CrossRef] [PubMed]
- Dri, P.; Gasparini, C.; Menegazzi, R.; Cramer, R.; Albéri, L.; Presani, G.; Garbisa, S.; Patriarca, P. TNF-Induced shedding of TNF receptors in human polymorphonuclear leukocytes: Role of the 55-kDa TNF receptor and involvement of a membrane-bound and non-matrix metalloproteinase. J. Immunol. 2000, 165, 2165–2172. [Google Scholar] [CrossRef] [Green Version]
- van Mierlo, G.J.; Scherer, H.U.; Hameetman, M.; Morgan, M.E.; Flierman, R.; Huizinga, T.W.; Toes, R.E. Cutting edge: TNFR-shedding by CD4+CD25+ regulatory T cells inhibits the induction of inflammatory mediators. J. Immunol. 2008, 180, 2747–2751. [Google Scholar] [CrossRef] [Green Version]
- Babic, A.; Shah, S.M.; Song, M.; Wu, K.; Meyerhardt, J.A.; Ogino, S.; Yuan, C.; Giovannucci, E.L.; Chan, A.T.; Stampfer, M.J.; et al. Soluble tumour necrosis factor receptor type II and survival in colorectal cancer. Br. J. Cancer 2016, 114, 995–1002. [Google Scholar] [CrossRef] [Green Version]
- Heemann, C.; Kreuz, M.; Stoller, I.; Schoof, N.; von Bonin, F.; Ziepert, M.; Löffler, M.; Jung, W.; Pfreundschuh, M.; Trümper, L.; et al. Circulating levels of TNF receptor II are prognostic for patients with peripheral T-cell non-Hodgkin lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3637–3647. [Google Scholar] [CrossRef] [Green Version]
- Tarhini, A.A.; Lin, Y.; Yeku, O.; LaFramboise, W.A.; Ashraf, M.; Sander, C.; Lee, S.; Kirkwood, J.M. A four-marker signature of TNF-RII, TGF-α, TIMP-1 and CRP is prognostic of worse survival in high-risk surgically resected melanoma. J. Transl. Med. 2014, 12, 19. [Google Scholar] [CrossRef] [Green Version]
- Warzocha, K.; Bienvenu, J.; Ribeiro, P.; Moullet, I.; Dumontet, C.; Neidhardt-Berard, E.M.; Coiffier, B.; Salles, G. Plasma levels of tumour necrosis factor and its soluble receptors correlate with clinical features and outcome of Hodgkin’s disease patients. Br. J. Cancer 1998, 77, 2357–2362. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998, 92, 4150–4166. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.Y.; Wang, G.X.; Yin, B.; Ozao, J.; Ku, T.; Divino, C.M.; Chen, S.H. Reversion of immune tolerance in advanced malignancy: Modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood 2008, 111, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004, 64, 6337–6343. [Google Scholar] [CrossRef] [Green Version]
- Ba, H.; Li, B.; Li, X.; Li, C.; Feng, A.; Zhu, Y.; Wang, J.; Li, Z.; Yin, B. Transmembrane tumor necrosis factor-α promotes the recruitment of MDSCs to tumor tissue by upregulating CXCR4 expression via TNFR2. Int. Immunopharmacol. 2017, 44, 143–152. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Auguste, P.; Fallavollita, L.; Wang, N.; Burnier, J.; Bikfalvi, A.; Brodt, P. The host inflammatory response promotes liver metastasis by increasing tumor cell arrest and extravasation. Am. J. Pathol. 2007, 170, 1781–1792. [Google Scholar] [CrossRef] [Green Version]
- Ham, B.; Wang, N.; D’Costa, Z.; Fernandez, M.C.; Bourdeau, F.; Auguste, P.; Illemann, M.; Eefsen, R.L.; Høyer-Hansen, G.; Vainer, B.; et al. TNF Receptor-2 Facilitates an Immunosuppressive Microenvironment in the Liver to Promote the Colonization and Growth of Hepatic Metastases. Cancer Res. 2015, 75, 5235–5247. [Google Scholar] [CrossRef] [Green Version]
- Milette, S.; Hashimoto, M.; Perrino, S.; Qi, S.; Chen, M.; Ham, B.; Wang, N.; Istomine, R.; Lowy, A.M.; Piccirillo, C.A.; et al. Sexual dimorphism and the role of estrogen in the immune microenvironment of liver metastases. Nat. Commun. 2019, 10, 5745. [Google Scholar] [CrossRef]
- Ivagnes, A.; Messaoudene, M.; Stoll, G.; Routy, B.; Fluckiger, A.; Yamazaki, T.; Iribarren, K.; Duong, C.P.M.; Fend, L.; Caignard, A.; et al. TNFR2/BIRC3-TRAF1 signaling pathway as a novel NK cell immune checkpoint in cancer. Oncoimmunology 2018, 7, e1386826. [Google Scholar] [CrossRef]
- Dobrzycka, B.; Terlikowski, S.J.; Kowalczuk, O.; Kinalski, M. Circulating levels of TNF-alpha and its soluble receptors in the plasma of patients with epithelial ovarian cancer. Eur. Cytokine Netw. 2009, 20, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhao, Z.; Zhao, N. Clinical implications of tumor necrosis factor receptor 2 in breast cancer. Oncol. Lett. 2017, 14, 2393–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.W.; Chen, Q.Q.; Cao, J.; Xu, L.Q.; Tang, X.; Wang, J.; Zhang, J.; Dong, L.X. Expression of tumor necrosis factor receptor 2 in human non-small cell lung cancer and its role as a potential prognostic biomarker. Thorac. Cancer 2019, 10, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.J.; Owens, D.M.; Stamp, G.; Arnott, C.; Burke, F.; East, N.; Holdsworth, H.; Turner, L.; Rollins, B.; Pasparakis, M.; et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med. 1999, 5, 828–831. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, M.; Okabe, S.; Marino, M.W.; Sakai, A.; Sueoka, E.; Fujiki, H. Essential role of tumor necrosis factor alpha (TNF-alpha) in tumor promotion as revealed by TNF-alpha-deficient mice. Cancer Res. 1999, 59, 4516–4518. [Google Scholar] [PubMed]
- Arnott, C.H.; Scott, K.A.; Moore, R.J.; Robinson, S.C.; Thompson, R.G.; Balkwill, F.R. Expression of both TNF-alpha receptor subtypes is essential for optimal skin tumour development. Oncogene 2004, 23, 1902–1910. [Google Scholar] [CrossRef] [Green Version]
- Ungewickell, A.; Bhaduri, A.; Rios, E.; Reuter, J.; Lee, C.S.; Mah, A.; Zehnder, A.; Ohgami, R.; Kulkarni, S.; Armstrong, R.; et al. Genomic analysis of mycosis fungoides and Sézary syndrome identifies recurrent alterations in TNFR2. Nat. Genet. 2015, 47, 1056–1060. [Google Scholar] [CrossRef]
- Zhao, T.; Li, H.; Liu, Z. Tumor necrosis factor receptor 2 promotes growth of colorectal cancer via the PI3K/AKT signaling pathway. Oncol. Lett. 2017, 13, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Onizawa, M.; Nagaishi, T.; Kanai, T.; Nagano, K.; Oshima, S.; Nemoto, Y.; Yoshioka, A.; Totsuka, T.; Okamoto, R.; Nakamura, T.; et al. Signaling pathway via TNF-alpha/NF-kappaB in intestinal epithelial cells may be directly involved in colitis-associated carcinogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G850–G859. [Google Scholar] [CrossRef]
- Suzuki, M.; Nagaishi, T.; Yamazaki, M.; Onizawa, M.; Watabe, T.; Sakamaki, Y.; Ichinose, S.; Totsuka, M.; Oshima, S.; Okamoto, R.; et al. Myosin light chain kinase expression induced via tumor necrosis factor receptor 2 signaling in the epithelial cells regulates the development of colitis-associated carcinogenesis. PLoS ONE 2014, 9, e88369. [Google Scholar] [CrossRef] [Green Version]
- Tummala, K.S.; Brandt, M.; Teijeiro, A.; Graña, O.; Schwabe, R.F.; Perna, C.; Djouder, N. Hepatocellular Carcinomas Originate Predominantly from Hepatocytes and Benign Lesions from Hepatic Progenitor Cells. Cell Rep. 2017, 19, 584–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Y.; Sun, K.; Liu, W.; Sheng, D.; Zhao, S.; Gao, L.; Wei, L. Tumor necrosis factor-α promotes hepatocellular carcinogenesis through the activation of hepatic progenitor cells. Cancer Lett. 2018, 434, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Zhao, Q.; An, L.; Jiao, S.; Li, R.; Sang, Y.; Liao, J.; Nie, P.; Wen, F.; Ju, J.; et al. A TNFR2-hnRNPK axis promotes primary liver cancer development via activation of YAP signaling in hepatic progenitor cells. Cancer Res. 2021, 81, 3036–3050. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.H.; Camargo, F.D.; Yimlamai, D. Hippo Signaling in the Liver Regulates Organ Size, Cell Fate, and Carcinogenesis. Gastroenterology 2017, 152, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Lucchini, F.; Sacco, M.G.; Hu, N.; Villa, A.; Brown, J.; Cesano, L.; Mangiarini, L.; Rindi, G.; Kindl, S.; Sessa, F.; et al. Early and multifocal tumors in breast, salivary, harderian and epididymal tissues developed in MMTY-Neu transgenic mice. Cancer Lett. 1992, 64, 203–209. [Google Scholar] [CrossRef]
- Sangaletti, S.; Tripodo, C.; Ratti, C.; Piconese, S.; Porcasi, R.; Salcedo, R.; Trinchieri, G.; Colombo, M.P.; Chiodoni, C. Oncogene-driven intrinsic inflammation induces leukocyte production of tumor necrosis factor that critically contributes to mammary carcinogenesis. Cancer Res. 2010, 70, 7764–7775. [Google Scholar] [CrossRef] [Green Version]
- Warren, M.A.; Shoemaker, S.F.; Shealy, D.J.; Bshar, W.; Ip, M.M. Tumor necrosis factor deficiency inhibits mammary tumorigenesis and a tumor necrosis factor neutralizing antibody decreases mammary tumor growth in neu/erbB2 transgenic mice. Mol. Cancer Ther. 2009, 8, 2655–2663. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Zhao, N.; Wu, N. TNFR2 promotes Adriamycin resistance in breast cancer cells by repairing DNA damage. Mol. Med. Rep. 2017, 16, 2962–2968. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yang, Y.; Yuan, F.; Huang, J.; Xu, W.; Mao, B.; Yuan, Z.; Bi, W. TNFα-YAP/p65-HK2 axis mediates breast cancer cell migration. Oncogenesis 2017, 6, e383. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Bhat, K.; Duhacheck-Muggy, S.; Ioannidis, A.; Zhang, L.; Nguyen, N.T.; Moatamed, N.A.; Pajonk, F. Tumor necrosis factor receptor signaling modulates carcinogenesis in a mouse model of breast cancer. Neoplasia 2021, 23, 197–209. [Google Scholar] [CrossRef]
- Medler, J.; Nelke, J.; Weisenberger, D.; Steinfatt, T.; Rothaug, M.; Berr, S.; Hünig, T.; Beilhack, A.; Wajant, H. TNFRSF receptor-specific antibody fusion proteins with targeting controlled FcγR-independent agonistic activity. Cell Death Dis. 2019, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Torrey, H.; Khodadoust, M.; Tran, L.; Baum, D.; Defusco, A.; Kim, Y.H.; Faustman, D.L. Targeted killing of TNFR2-expressing tumor cells and T(regs) by TNFR2 antagonistic antibodies in advanced Sézary syndrome. Leukemia 2019, 33, 1206–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guéry, L.; Dubrot, J.; Lippens, C.; Brighouse, D.; Malinge, P.; Irla, M.; Pot, C.; Reith, W.; Waldburger, J.M.; Hugues, S. Ag-presenting CpG-activated pDCs prime Th17 cells that induce tumor regression. Cancer Res. 2014, 74, 6430–6440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moseman, E.A.; Liang, X.; Dawson, A.J.; Panoskaltsis-Mortari, A.; Krieg, A.M.; Liu, Y.J.; Blazar, B.R.; Chen, W. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. J. Immunol. 2004, 173, 4433–4442. [Google Scholar] [CrossRef] [Green Version]
- Nie, Y.; He, J.; Shirota, H.; Trivett, A.L.; Yang, D.; Klinman, D.M.; Oppenheim, J.J.; Chen, X. Blockade of TNFR2 signaling enhances the immunotherapeutic effect of CpG ODN in a mouse model of colon cancer. Sci. Signal. 2018, 11, eaan0790. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, K.C.; Pinckard, J.K.; Arthur, C.D.; Dehner, L.P.; Goeddel, D.V.; Schreiber, R.D. Monoclonal antibodies specific for murine p55 and p75 tumor necrosis factor receptors: Identification of a novel in vivo role for p75. J. Exp. Med. 1995, 181, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.S.; Mistry, B.; Guillard, S.; Ulrichsen, J.C.; Sandercock, A.M.; Wang, J.; González-Muñoz, A.; Parmentier, J.; Black, C.; Soden, J.; et al. Phenotypic screening reveals TNFR2 as a promising target for cancer immunotherapy. Oncotarget 2016, 7, 68278–68291. [Google Scholar] [CrossRef] [Green Version]
- Mårtensson, L.; Kovacek, M.; Holmkvist, P.; Semmrich, M.; Svensson, C.; Blidberg, T.; Carl Roos, C.; McAllister, A.; Demiri, M.; Borggren, M.; et al. 725 Pre-Clinical Development of TNFR2 Ligand-Blocking BI-1808 for Cancer Immunotherapy. J. Immuno Ther. Cancer 2020, 8, A768. [Google Scholar] [CrossRef]
- Tam, E.M.; Fulton, R.B.; Sampson, J.F.; Muda, M.; Camblin, A.; Richards, J.; Koshkaryev, A.; Tang, J.; Kurella, V.; Jiao, Y.; et al. Antibody-mediated targeting of TNFR2 activates CD8(+) T cells in mice and promotes antitumor immunity. Sci. Transl. Med. 2019, 11, eaax0720. [Google Scholar] [CrossRef]
- Chen, Y.; Jia, M.; Wang, S.; Xu, S.; He, N. Antagonistic Antibody Targeting TNFR2 Inhibits Regulatory T Cell Function to Promote Anti-Tumor Activity. Front. Immunol. 2022, 13, 835690. [Google Scholar] [CrossRef]
- Filbert, E.; Krishnan, S.; Alvarado, R.; Huang, G.; Bahjat, F.; Yang, X. 693 APX601, a Novel TNFR2 antagonist antibody for cancer immunotherapy. J. Immuno Therapy Cancer 2020, 8, A417. [Google Scholar] [CrossRef]
- Krishnan, S.; Alvarado, R.; Huang, G.; Yang, X.; Filbert, E.L. Abstract LB175: APX601, a Potent TNFR2 Antagonist as a Novel and Promising Approach to Reverse Tumor Immune Suppression. Cancer Res. 2021, 81, LB175. [Google Scholar] [CrossRef]
- Mårtensson, L.; Cleary, K.; Semmrich, M.; Kovacek, M.; Holmkvist, P.; Svensson, C.; Demiri, M.; Blidberg, T.; Thornberg, U.-C.; Pitic, V.; et al. Abstract 936: Targeting TNFR2 for cancer immunotherapy: Ligand blocking depletors versus receptor agonists. Cancer Res. 2020, 80, 936. [Google Scholar] [CrossRef]
- Wei, S.; Fulton, R.; Lu, Y.-Y.; Zhang, Q.; Zhou, H.; Raue, A.; Chen, M.; Xu, W.; Cai, X.; Crivello, J.; et al. Abstract 1883: Mechanism of action and biomarker strategy for HFB200301, an anti-TNFR2 agonist antibody for the treatment of cancer. Cancer Res. 2021, 81, 1883. [Google Scholar] [CrossRef]
- Sum, C.S.; Danton, M.; hu, Q.; Pritsker, A.; Lin, R.; Yu, R.; Chen, K.; Tang, F.; Pohl, T.; Wallner, S.; et al. Abstract 1869: Novel TNFR2 antibodies to overcome T cell exhaustion and suppressive tumor microenvironment. Cancer Res. 2021, 81, 1869. [Google Scholar] [CrossRef]
- Richards, J.; Wong, C.; Koshkaryev, A.; Fulton, R.; Camblin, A.; Sampson, J.; Luus, L.; Suchy, J.; Grabow, S.; Kurella, V.; et al. Abstract 4846: MM-401, a novel anti-TNFR2 antibody that induces T cell co-stimulation, robust anti-tumor activity and immune memory. Cancer Res. 2019, 79, 4846. [Google Scholar] [CrossRef]
- Sampson, J.F.; Kurella, V.B.; Paragas, V.; Kumar, S.; Lulo, J.E.; Qiu, J.A.; Razlog, M.; Fulton, R.B.; Camblin, A.J.; Richards, J.M.; et al. Abstract 555: A novel human TNFR2 antibody (MM-401) modulates T cell responses in anti-cancer immunity. Cancer Res. 2019, 79, 555. [Google Scholar] [CrossRef]
- Case, K.; Tran, L.; Yang, M.; Zheng, H.; Kuhtreiber, W.M.; Faustman, D.L. TNFR2 blockade alone or in combination with PD-1 blockade shows therapeutic efficacy in murine cancer models. J. Leukoc. Biol. 2020, 107, 981–991. [Google Scholar] [CrossRef]
- Jiang, M.; Liu, J.; Yang, D.; Tross, D.; Li, P.; Chen, F.; Alam, M.M.; Faustman, D.L.; Oppenheim, J.J.; Chen, X. A TNFR2 antibody by countering immunosuppression cooperates with HMGN1 and R848 immune stimulants to inhibit murine colon cancer. Int. Immunopharmacol. 2021, 101, 108345. [Google Scholar] [CrossRef]
- Vredevoogd, D.W.; Kuilman, T.; Ligtenberg, M.A.; Boshuizen, J.; Stecker, K.E.; de Bruijn, B.; Krijgsman, O.; Huang, X.; Kenski, J.C.N.; Lacroix, R.; et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell 2019, 178, 585–599.e515. [Google Scholar] [CrossRef]
- Aguadé-Gorgorió, J.; McComb, S.; Eckert, C.; Guinot, A.; Marovca, B.; Mezzatesta, C.; Jenni, S.; Abduli, L.; Schrappe, M.; Dobay, M.P.; et al. TNFR2 is required for RIP1-dependent cell death in human leukemia. Blood Adv. 2020, 4, 4823–4833. [Google Scholar] [CrossRef] [PubMed]
- Govindaraj, C.; Madondo, M.; Kong, Y.Y.; Tan, P.; Wei, A.; Plebanski, M. Lenalidomide-based maintenance therapy reduces TNF receptor 2 on CD4 T cells and enhances immune effector function in acute myeloid leukemia patients. Am. J. Hematol. 2014, 89, 795–802. [Google Scholar] [CrossRef] [PubMed]
- van der Most, R.G.; Currie, A.J.; Mahendran, S.; Prosser, A.; Darabi, A.; Robinson, B.W.; Nowak, A.K.; Lake, R.A. Tumor eradication after cyclophosphamide depends on concurrent depletion of regulatory T cells: A role for cycling TNFR2-expressing effector-suppressor T cells in limiting effective chemotherapy. Cancer Immunol. Immunother. 2009, 58, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Li, R.; Hu, H.; Hu, Y.; Chen, X. Modulation of Regulatory T Cell Activity by TNF Receptor Type II-Targeting Pharmacological Agents. Front. Immunol. 2018, 9, 594. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| TNFR2 Expressing Cell Type | Effect on Cell | Effect on Tumor | Ref. |
|---|---|---|---|
| MDSC | CXCR4 expression and tumor recruitment Increase in suppressive activity | Immune escape Metastasis | [81,89,125,128] |
| Treg | Expansion Phenotypic stability Increase in suppressive activity | Immune escape Metastasis | [59,98] |
| NK cell | Inhibition of NKp46 expression | Metastasis | [130] |
| Tumor cell | Survival signaling Proliferation | Tumor progression Malignant transformation | [105,106,107,137,143,145,146,147] |
| Tumor cell | Cell-death sensitization | Anti-tumoral | [110,111] |
| Antibody | Company | Mode of Action | Status | Relevant Patent | Ref. |
|---|---|---|---|---|---|
| AN3025 | Adlai Norty USA Inc. | Antagonist ADCC | Preclinical | - | [160] |
| APX601 | Apexigen Inc. | Antagonist ADCC | Preclinical | WO2021/055253A2 | [161,162] |
| Bi-1808 | BioInvent International AB | Antagonist | NCT04752826 | WO2020/089474A1 | [158] |
| Bi-1910 | BioInvent International AB | Agonist | Preclinical | WO2020/089473A2 | [163] |
| BITR2101 | Boston Immune Technologies & Therapeutic Inc. | - | Preclinical | - | - |
| HFB200301 | HiFiBiO Therapeutics | Agonist | NCT05238883 | WO2021/141907A1 | [164] |
| LBL-019 | Nanijng Leads Biolabs Co Ltd. | - | NCT05223231 | WO2021/249542A1 | - |
| NBL-020 | NovaRock Biotherapeutics | FcγR dependent | Preclinical | - | [165] |
| MM-401 | Merrimack Pharmaceuticals | Agonist ADCC | Preclinical | WO2020/180712A1 WO2020/061210A1 | [166,167] |
| SIM0235 | Simcere | Antagonist ADCC | Preclinical | WO2021/023089 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medler, J.; Kucka, K.; Wajant, H. Tumor Necrosis Factor Receptor 2 (TNFR2): An Emerging Target in Cancer Therapy. Cancers 2022, 14, 2603. https://doi.org/10.3390/cancers14112603
Medler J, Kucka K, Wajant H. Tumor Necrosis Factor Receptor 2 (TNFR2): An Emerging Target in Cancer Therapy. Cancers. 2022; 14(11):2603. https://doi.org/10.3390/cancers14112603
Chicago/Turabian StyleMedler, Juliane, Kirstin Kucka, and Harald Wajant. 2022. "Tumor Necrosis Factor Receptor 2 (TNFR2): An Emerging Target in Cancer Therapy" Cancers 14, no. 11: 2603. https://doi.org/10.3390/cancers14112603