1. Introduction
Prostate cancer (PC) is the most commonly occurring cancer and the second leading cause of cancer mortality in men in the United States. Approximately 248,530 men were diagnosed with PC and approximately 34,130 men died in the US in 2021 [
1]. In patients with primary prostate cancer, bone metastases are by far the predominant metastatic site and the primary contributing factor to this mortality. In a study that performed autopsies in 1589 patients who died with metastatic castrate resistant prostate cancer, bone metastases were found in 90% confirming this dominant issue [
2]. In metastatic prostate cancer there is a nearly 90% response to androgen deprivation therapy. Although there are currently multiple therapies targeting the androgen receptor, there is a universal disease relapse despite the encouraging and prolonged initial tumor responses [
3]. Prostate cancer stem cells are believed to be integral to this ultimate disease progression. Cancer stem cells (CSCs) are a small subpopulation of cells that possess characteristics associated with normal stem cells, allowing for self-renewal and differentiation. They are integrally responsible for tumor relapses as well as the development of therapeutic resistance and may be protected by the bone marrow niche [
4,
5]. The bone marrow niche plays a significant role in promoting prostate cancer cell growth in the bone marrow and this relationship with the microenvironment appears to contribute to the treatment resistance of CSCs, [
6]. Despite our increasing knowledge of the biology and role of CSCs and their interaction with the tumor stroma, therapeutic options which specifically target CSC are limited.
Using a novel inhibitor, we evaluated mitochondria-associated granulocyte–macrophage colony-stimulating factor signaling molecule (Magmas) as a potential therapeutic target in prostate cancer. Magmas, a highly conserved and essential protein overexpressed in aggressive prostate cancer, is an attractive novel target. Mitochondrial targets such as Magmas have been shown to have an increasing role in the development of cancer stem cells and therapeutic resistance [
7,
8]. Traditional chemotherapy is dependent on cell growth to be effective and the dormancy of CSCs is one of the reasons for their treatment resistance. Mitochondrial-targeted therapy, however, does not have this dependence on cell turnover.
Magmas was first identified in a study investigating granulocyte colony-stimulating factor (GM-CSF)-responsive genes. In that study, rapid induction of Magmas occurred when the human myeloid cell line PGMD1 was exposed to GM-CSF when compared to interleukin-3 [
9]. Subsequent studies characterized its gene structure, confirmed its mitochondrial location, and identified its potential role in protein transport and mitochondrial biogenesis [
10]. Further contributing to the knowledge and function of Magmas were experiments showing that it is overexpressed in two-thirds of pituitary adenomas. It is also highly expressed in two ACTH-secreting pituitary cell lines and loss-of-function studies demonstrated the role of Magmas in protecting these cells from apoptosis, signifying a role in cancer development [
11,
12]. One study examined Magmas expression by immunohistochemistry in human prostate cancer tissues obtained at the time of radical prostatectomy. Magmas was expressed in 14 out of 15 tumors, with variable expression ranging from one tumor demonstrating weak staining in 10% of tumor cells to another showing strong staining in greater than 90% of malignant cells. Normal prostate showed either no or only weak staining in a small percentage of cells whereas the premalignant lesion, prostatic intraepithelial neoplasia (PIN), had a staining pattern intermediate between the normal and malignant tissues [
13]. Although this data supports the potential role of Magmas in the carcinogenic process, its role in the development of treatment resistance is still largely unknown.
As previously reported, we designed several small-molecule Magmas inhibitors (SMMI) and assayed for their effects on proliferation in yeast [
14]. The most active compound, Bhaskar Technology #9(BT#9), inhibited growth at a 4 μM scale and this compound was shown by fluorometric titration to bind to Magmas with a K
d = 33 μM. In this this prior study, target specificity of BT#9 was established by demonstrating direct binding of the compound to Magmas [
14]. Here, we evaluated Magmas expression in human prostate cancer cell lines, patient tissues, and publicly available prostate cancer patient datasets and further elucidated the anticancer effect of BT#9 on prostate cancer cells in vitro. Our specific objectives were to establish Magmas as a potential target in prostate cancer, to further delineate the mechanism of BT#9 and to investigate it as a potential target in treatment-resistant prostate cancer cells.
2. Materials and Methods
2.1. Chemicals and Reagents
BT#9 was synthesized using our previous established procedures [
14]. Docetaxel, N-acetyl-l-cysteine, were purchased from Sigma (Burlington, MA, USA). Z-VAD-FMK was purchased from R&D system (Minneapolis, MN, USA). TRAIL and SMAC mimic (Nirinapanit) were purchased from PeproTech (Rocky Hill, NJ, USA) and ApexBio Tech LLC (Houston, TX, USA). MTS and PMS were purchased from Abcam (Waltham, MA, USA) and Sigma (Burlington, MA, USA). Most chemicals were dissolved in dimethyl sulfoxide (DMSO) at proper stocking solution (1000-fold of working concentration), the DMSO volume in final working concentration was equal to or below 0.1% (
V/
V) and had no noticeable inhibitory effects on cultured cells.
2.2. Cell Culture
Human prostate cancer cell lines LNCaP, PC3, DU145 and the EBV transformed prostate/stromal cell line WPMY-1 were acquired from American Type Culture Collection (ATCC, Manassas, VA, USA; catalog numbers CRL-1740, CRL-1435, HTB-81 and CRL-2854, respectively). The cell lines PC3, DU145 and WPMY-1 were maintained in Dulbecco modified Eagle medium (DMEM). RPMI-1640 medium was used for LNCaP cells. In all cases, the medium was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin solution.
2.3. Patient Tissues
Human prostate tissue samples (normal adjacent and prostate tumor tissue pairs) were purchased from the Biospecimen Repository Core Facility, University of Kansas Cancer Center. The Institutional Review Board (IRB) of the Veterans Administration Medical Center, Kansas City gave approval for the use of the tissue samples purchased under the protocol named: “Effect of Magmas inhibitor in Xenograft mouse model”, ID: 00835.
2.4. Western Blotting
Protein was extracted from cells growing on 6-well plates with or without treatment with BT#9. Cell and tissue lysates were prepared in the lysis buffer (50 mm Tris-HCl, pH 8.0, 0.1% SDS, 150 mm NaCl, 1% Nonidet P-40, protease inhibitor mixture: 1 μg/mL aprotinin, 1 μg of leupeptin, and 1.0 mm PMSF). Equal amounts of protein were loaded to 10% SDS-PAGE and transferred to a nitrocellulose membrane by tank blotting system (Bio-Rad, Hercules, CA, USA). Membranes were incubated with anti-Magmas antibody overnight followed by HRP-conjugated secondary antibodies. The expression of the protein, β-actin, was used as a loading control. The polyclonal anti-Magmas antibody was a kind gift from Paul T. Jubinsky, M.D., Ph.D. and Mary Short, Ph.D., Albert Einstein Medical College, New York. The Caspase-3, Anti-β-actin, the Anti-rabbit IgG, HRP-linked and Anti-mouse IgG, HRP-linked antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA); catalog numbers 9669, 3700, 7074 and 7076, respectively. Thermo Scientific 1-Step Ultra TMB-Blotting Solution (catalog number # 37574) was used to determine the protein band intensity.
2.5. MTT and MTS Cell Viability Assays
MTT assay was performed using the Vybrant
® MTT Cell viability Assay Kit purchased from Life Technologies (Grand Island, NY, USA, catalogue number: V13154) according to manufacturer’s protocol. Briefly, the cells were seeded in a 96-well plate at 10,000/well and underwent overnight incubation, then treated with 1–20 μM of BT9 for 24 and 48 h. MTT reagent was then added to the cells and the absorbance of the color developed was measured at 570 nm. The Statistics Calculators (
www.danielsoper.com) was used to analyze the data. In addition, for direct comparison between cell viability and apoptosis rate from the same size plate, or for further validation purposes, we also ran MTS Cell Viability Assay for cell cultured on 6- or 12-well plates; the optical density at 490 nm was read by Bioteck SynergyǁMulti-Detection Microplate Reader (Winooski, VT, USA).
2.6. Terminal Deoxynucleotidyl Transferase (TdT) dUTP Nick-End Labeling (TUNEL) Assay
Late stage of apoptosis was quantified by TUNEL based Apo-BRDU kit (BD Biosciences, San Jose, CA, USA). Briefly, collected cells were fixed with 1% paraformaldehyde for 30 min on ice. Cells were then resuspended in ice-cold 70% ethanol and stored at −20 °C until use. Fragmented DNA was labeled by the staining buffer, including fluorescein-dUTP and TdT, for 90 min at 37 °C. After washing and centrifuge, the cell pellet was resuspended in 0.5 mL of PBS containing 5 µg/mL propidium iodide and 500 µg/mL RNase A. After 30 min of incubation in the dark at room temperature, cells were analyzed using a FACScan LSR II (BD Biosciences, San Jose, CA, USA).
2.7. Detection of Mitochondrial Membrane Potential (ΔΨm) by Flow Cytometry
Mitochondrial depolarization was monitored with the potentiometric dye JC-1 using the Mitoprobe assay kit (Invitrogen) according to the manufacturer’s instructions. Cells were treated with BT#9 for 4 and 24 h, then incubated in medium containing 2 µM JC-1 for 10 min. The samples were washed with PBS, resuspended in 500 μL PBS, and kept on ice until analyzed by BD LSR II cytometer. Results were further processed with Flowjo Software (Tree Star, Inc., Ashland, OR, USA). JC-1 is readily taken up by cells and healthy mitochondria, and the JC-1 probe (green fluorescence) is a monomer at low membrane potential but yields ‘Jaggregates’ (red fluorescence) at elevated membrane potential. The mitochondrial membrane potential was evaluated as the red/green fluorescence intensity ratio.
2.8. Immunofluorescence and Fluorescence Microscopy
Cells were grown on 4-well chamber slide (NEST Scientific, Woodbridge Township, NJ, USA) and fixed with 4% formaldehyde at room temperature for 15 min. After rinsing three times, slides were blocked by blocking buffer at room temperature for 60 min. Next, they were incubated with cleaved caspase-3 antibody (Cell Signaling Technology, Danvers, MA, USA)—conjugated Alexa Fluor 488 for 1 h at RT. Pictures were taken by Olympus (Tokyo, Japan) fluorescence microscope system, including CKX41 inverted microscope, DP74 camera and CellSense entry Software.
2.9. Annexin V/SYTOX Green Assay for Apoptosis and Necrosis
Apoptotic and necrosis was determined using the Dead Cell Apoptosis Kit with Annexin V- APC and SYTOX® Green for flow cytometry (Life Technologies, Grand Island, NY, USA; catalogue number: V35113) according to manufacturer’s protocol. Briefly, apoptotic cells were detected by the binding of annexin V to externalized phosphatidylserine. Annexin V+/SYTOX Green- were considered early apoptotic death. The late apoptotic and necrotic cells have non-intact membranes that permit SYTOX® Green stain entrance to cellular nucleic acids. These populations of cells were detected using the Flow Cytometry Core Laboratory at The University of Kansas Medical Center.
2.10. Detection of Mitochondria and Intracellular ROS Production
MitoSOX red mitochondrial superoxide indicator (Invitrogen, Waltham, MA, USA) was used to measure the presence of mitochondria superoxide. The procedure was conducted according to the manual with slight modification. Before use, MitoSOX Red reagent was dissolved in DMSO 13 µL to yield a 5 mM stock solution. After BT#9 treatment at desired time, 1 μL of stocking solution was added to medium of 100 µL in an Eppendorf tube and vortexed, then added directly to 1 mL medium in a 12-well plate with a final working concentration of 2.5 μM. After incubation at 37 °C for 20 min, analysis was conducted by both fluorescence microscopy and BD LSR II cytometer (green laser with emission wavelength at 575 nm, PE channel). In addition, analysis of intracellular ROS generation was evaluated by adding a fluorescent CellROX Green Reagent (Invitrogen, Waltham, Massachusetts), followed by flow fluorescence microcopy and cytometry. Briefly, cells were plated in 12-well plates at a final density of 2 × 105 cells/well and incubated overnight. Cells were then treated with the BT#9 at concentration of 10 µM or 20 µM for 24 h. Cells were stained with 2.5 μM CellROX Green Reagent for 20 min at cell culture incubator. Finally, analysis was conducted by both fluorescence microscopy and BD LSR II cytometer (blue laser with emission wavelength at 515 nm, FITC channel).
2.11. Dataset Analysis
Prostate cancer patient datasets available in the Oncomine and cBioPortal databases were queried for Magmas (TIMM16, PAM16) mRNA overexpression and copy number alterations, respectively.
2.12. Drug Interaction Relationships Analysis
Drug combination effects were determined using Compusyn analytical (version 1.0) software (T. C. Chou and N. Martin, Memorial Sloan-Kettering Cancer Center, New York and were expressed as a combination index (CI). CI is a quantitative measure of the degree of drug interaction. CI < 0.9, CI = 0.9–1.1, or CI > 1.1 represents synergistic, additive, and antagonistic relationships, respectively.
2.13. Statistical Analysis
Data are expressed as the mean ± standard deviation. All statistical analyses were performed using GraphPad Prism software (version 7.0, La Jolla, CA, USA). One-way analysis of variance and Tukey’s tests were performed for multiple group comparisons. * p < 0.05 was considered statistically significant.
4. Discussion
Mitochondria play a critical role in cancer bioenergetics and are considered potential targets for cancer treatment [
32]. Current mitochondria-targeting anticancer drugs still have limited efficacy, so it is imperative to develop novel and potent mitochondria-targeting agents. Based on the reported observations that Magmas may be a viable cancer therapeutic target, we developed the first reported Magmas inhibitor, BT#9 [
14], which is now being validated by other investigators in other malignances [
33,
34], but our report is the first exploring the effect of BT#9 on prostate cancers. MTT assays found that BT#9 reduced significant cell viability in multiple prostate cancer cell lines including PC3, DU145 and LNCaP in a dose-dependent and time-dependent manner, but interestingly, BT#9 spared normal prostate cancers WPMY-1. The underlying mechanism might be contributed to lower expression of Magmas in normal WPMY-1 cells. In terms of the specific binding of BT#9, it was reported that BT#9 can bind Magmas protein as fluorometric titration and molecular modeling has shown [
14]. Our protein expression analysis by Western blot further confirmed that the Magmas protein was decreased dramatically by BT#9 at the concentration of 10 µM but not 1 µM after 24 h. The insufficient downregulation of Magmas protein by 1 µM BT#9 is consistent with insignificantly reduced cell viability at 24 h.
Mitochondria is not only indispensable for energy production of cancer cells, but is also the critical regulator of the mitochondrial pathway of apoptosis. In view of the intra-mitochondria location of Magmas and its role of apoptosis inhibition as mentioned before, we initially expected that targeting mitochondria protein Magmas can induce predominant apoptotic cell death via mitochondria apoptosis, which is the tentative main mode of cell death of prostate cancer cells after treatment with BT#9 at the concentration of 10 or 20 µM. Unexpectedly, when applied pan-caspase inhibitor Z-VAD-FMK, the 10 or 20 µM BT#9 treated cell death was not protected significantly. Therefore, we presume that activation of caspase or apoptosis induction might not be a critical event for cell death induced by BT#9.
It is widely accepted that under the cell death scenario, that the loss of MMP is a hallmark of intrinsic apoptosis and an upstream event of caspase activation in the intrinsic pathway. JC-1 is considered the gold standard for the evaluation of the loss of MMP. We ran JC-1 fluorescence assay and detected decreased red/green fluorescence ratio as earlier as 4 h after BT#9 (20 μM) treatment, which is a sign of decreased MMP and an early event of apoptosis activation. In addition, the loss of MMP is presumed to be accompanied with the opening of the mitochondrial permeability transition pore (MPTP), allowing small molecules (such as cytochrome c) to be released into the cytosol where caspases will be activated. Caspase-3 is the main executive caspase in apoptosis. In our study, both Western blot and immunofluorescence microscopy demonstrated limited activation of caspase-3, which explained why the caspase inhibitor failed to protect BT#9 induced cell death. In addition, Z-VAD-FMK only inhibits multiple caspases activation, but it may not inhibit caspase-independent apoptosis. The concept of caspase independent apoptosis is still under debate. In spite of this uncertainty, we needed to identify and quantify the apoptosis rate by the TUNEL based apoptosis assay, which exclusively detects the characteristic event in later stages of apoptosis.
Necrosis has been classically defined as an unprogrammed form of cell death that occurs in response to overwhelming chemical or physical insult. Fluorescence microscopy after PI staining proved dramatic cell death 48 h after BT#9 treatment (
Figure 4F), and Anexin V assay (
Figure 4G) further proved the cell death was a mode of more necrosis less apoptosis. To sum up, BT#9 caused significant cytotoxicity, might devastate cancer cells and mainly caused necrosis, while apoptosis was only activated to a low level due to an unknown suppressed mechanism.
As mentioned earlier, Srivastava et al. [
29] have shown that Magmas may function as a regulator of ROS and protects cells against oxidative stress-mediated damages. Magmas protein promotes cellular tolerance toward oxidative stress by enhancing antioxidant enzyme activity, thus preventing the induction of apoptosis. Investigators in their study showing siRNA-mediated knockdown of Magmas resulted in a more than 1.5-fold increase in ROS levels, while overexpression of Magmas rescues the viability of these cancer cells from oxidative stress. Therefore, we intended to observe and measure the mitochondrial and intercellular ROS after BT#9 treatment. In this study, we found generation of ROS after BT#9 treatment detected by two ROS detection assays which compensate and validate each other. The first one detects mitochondria ROS but is limited to superoxide only, the second one detects intracellular total ROS but cannot distinguish an intra-mitochondria or extra-mitochondria source. When results are combined, we conclude that BT#9 definitely induced intracellular ROS accumulation, with a possibility of BT#9 targeting mitochondria and inducing intra-mitochondria ROS initially. Because in our dynamic observation, we found the intra-mitochondrial fluorescence signal reached the strongest signal around 8 h as we selected this peak time for data record, while the strongest signal intracellularly signal was around 24 h. Under the same experimental condition, NAC in combination not only suppresses ROS accumulation remarkably but also significantly protected BT#9 induced cell death, indicating the pivotal role of ROS in BT#9 induced cell death. The dramatic abrogation of cell death in prostate cancer cells treated by BT#9 suggests that ROS is a possible mediator of cell death. We also found that the intracellular ROS induced by BT#9 at the concentration lower than 10 μΜ is limited, which is proportional to weak cytotoxicity measured by either MTT or MTS. This provides another support for the notion that the ROS-dependent pathway is a critical mechanism of the anti-cancer effect of BT#9.
Numerous reports showed cancer cells exhibit a higher ROS level than normal cells for metabolic purposes, potential anticancer therapy can kill cancer cells by pushing cancer cells beyond a breaking point where the intracellular ROS level can damage cells. However, the death mode, conduct pathway and detailed mechanism of ROS-induced cell death is still not fully understood. What we know is that under different scenarios, ROS may induce apoptosis, necrosis, autophagy or mixed modes of cell death, depending on the cell death trigger, cell type and many other conditions. In this study, ROS not only induced cell apoptosis, but more importantly necrosis. As some research has shown [
35], higher level of ROS may paradoxically inhibit the function of caspase and apoptosis. This finding may partly explain why apoptosis was not predominant in BT#9 induced cell death. Indeed, the mode of cell death still needs further investigation.
As mentioned earlier, Magmas protected ACTH-secreting pituitary adenoma from apoptotic stimuli such as staurosporine [
11], indicating its negatively regulating role in response to pro-apoptotic stimuli. As a Magmas inhibitor, we also hypothesized that BT#9 might act as “apoptosis regulator” rather than “apoptosis trigger”. So far, there is no report about the combination of BT#9 and any anticancer treatments; whether synergism exist or not is unknown. TRAIL selectively induces apoptotic cell death in a variety of cancer cells and is regarded as a therapeutic apoptosis inducing treatment in some clinic trials, but TRAIL resistance is prevalent in many solid cancers including prostate cancer. Therefore, we firstly tested the combination of TRIAL and sub-effective concentration of BT#9. Results of calculated CI value (0.22–0.72) proved that a slight synergistic effect existed, while positive control SMAC mimic has a much stronger synergistic effect on TRAIL. In clinic, docetaxel retains a key role in the treatment for metastatic hormone-refractory prostate cancer (HRPC), however, almost all patients develop drug resistance after treatment. We examined the combination effect between docetaxel and BT#9 but a CI of around 1 proved the combination is just additive. Interestingly, we did not prove a synergistic effect between docetaxel and SMAC mimic either, possibly because SMAC mimic is a potent apoptosis regulator but docetaxel-induced cell death might not be apoptosis-predominant at the tested doses, which may also explain why BT#9 synergized with TRAIL but not docetaxel in concurrent treatment. As mentioned earlier, the Magmas protein was originally identified as a protein involved in GM-CSF signaling [
9], we found that Magmas was also induced by anticancer treatment and other cell stress conditions. Western blot found Magmas protein in PC-3 cells was significantly upregulated by treatment with 1 μΜ docetaxel at 24 h and 48 h. It is too early to ascertain whether this inducible expression of Magmas is involved in the mechanism of drug resistance, but we did evaluate the sequential treatment of docetaxel and BT#9. We did not find a synergistic effect between docetaxel (0.1 or 1 μM) and BT#9 (1 or 2 μM), which is similar to the finding with concurrent treatment. Interestingly, there is slight synergistic effect when BT#9 is dosed at 5 μM as
Figure 6E shows. The molecular mechanism for this synergism needs further investigation.
Figure 7 summarizes the potential molecular mechanism of prostate cancer cell death induced by BT#9. In brief, BT#9 has an on-target effect on the function and the expression level of Magmas protein, followed by mitochondria disturbance. Both effects further cause ROS accumulation and then activate caspase-independent necrosis, which is the predominant mode of cell death after BT#9 treatment. In addition, mitochondrial disturbance causes intrinsic apoptosis but not dramatically possibly because caspase was suppressed by excess ROS paradoxically. In addition, ROS accumulation and mitochondrial disturbance may affect each other. Further studies are being planned to understand BT#9 mechanisms of prostate cancer cell death. Regardless, BT#9 is a potent Magmas inhibitor because it caused significant cell death at the concentration of 10 µM-20 µM in vitro. The real potential clinical application is to target treatment-resistant prostate cancer cells, including prostate cancer stem cells (PCSC). Mitochondria are the most prominent source of intracellular reactive oxygen species (ROS) and low levels of ROS have been implicated in cancer cell stemness. Mitochondrial-targeted redox-active agents [
32] may therefore provide a novel strategy to selectively target prostate cancer stem cells, with BT#9 being a potential candidate.
Two other published studies have evaluated BT#9 in other malignancies, one being ovarian carcinoma. Investigators found that BT#9, reduced the viability of an ovarian carcinoma carboplatin-resistant cell line (OV9) significantly more than the parental OV90 cell line, suggesting a potential role for Magmas inhibition in overcoming chemotherapeutic resistance [
33]. Their data have also shown that ovarian cancer cells after treatment with chemotherapy are enriched in a population of cells having CSC-like characteristics. Magmas overexpression has also been reported in human gliobastoma (GBM) resection samples and in tumors derived from the syngenetic subcutaneous injection of the GL261 murine glioblastoma cell line [
34]. In these experiments, BT#9 decreased proliferation in human GBM cell lines (D-54 MG, U-251 MG), murine embryonal stem cell lines (1123 Mes, 83 Mes) and a human glioblastoma stem cell line (HuPuP01). These published studies further support that Magmas inhibition is a viable treatment strategy, targeting both chemotherapeutic resistant and cancer-like stem cells.
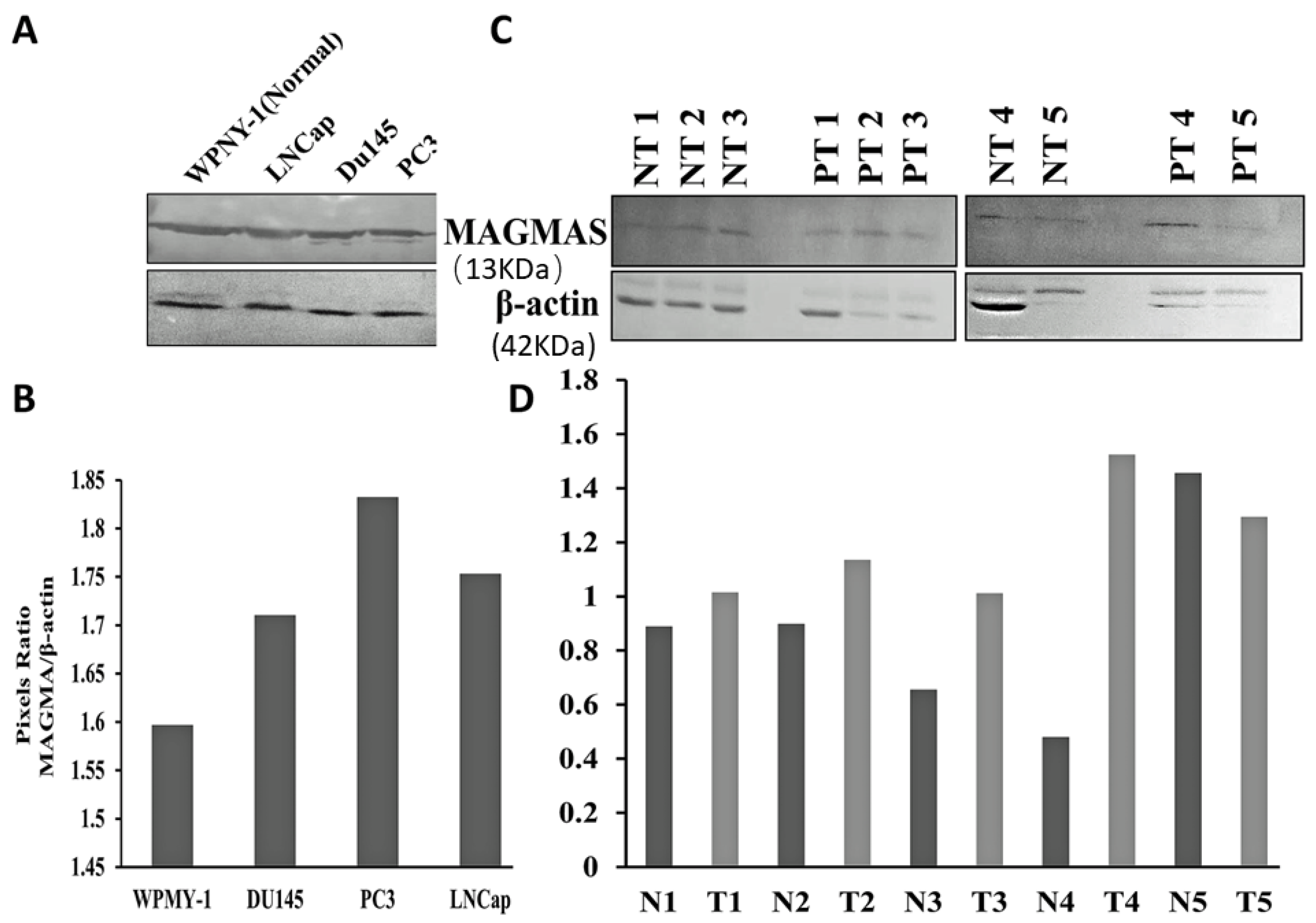
Our experiments were designed to determine whether Magmas inhibition has a potential role in the treatment of prostate cancer. We initially verified Magmas protein expression, which was present in both the WPMY-1 (the EBV transformed prostate/stromal cell line) used as control and the prostate cancer cell lines. The expression, however, was less in the EBV transformed prostate/stromal cell line. We then analyzed the Magmas protein expression in fresh adjacent normal prostate tissue and prostate tumor samples. Although the differences in expression were variable, in four of five samples, the prostate cancer adjacent normal tissues showed decreased expression of Magmas protein relative to the prostate cancer. With this method of tissue collection, prostate tissues neighboring the area of the tumor frequently contain premalignant changes, which may increase Magmas expression thereby impacting the magnitude of difference. Future human studies are needed to differentiate the change in Magmas expression across the spectrum of a normal prostate gland to glands affected purely by benign prostate hyperplasia, premalignant change, and metastatic disease, and further determine how expression changes with the development of treatment resistance.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}