
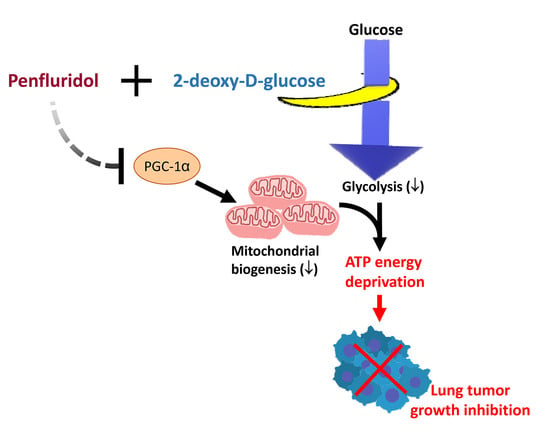
Synergistic Tumor Inhibition via Energy Elimination by Repurposing Penfluridol and 2-Deoxy-D-Glucose in Lung Cancer


 , and
, and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents, Chemical Inhibitors, and Antibodies
2.2. Cell Lines and Cell Culture
2.3. Cell Viability and Clonogenic Assays
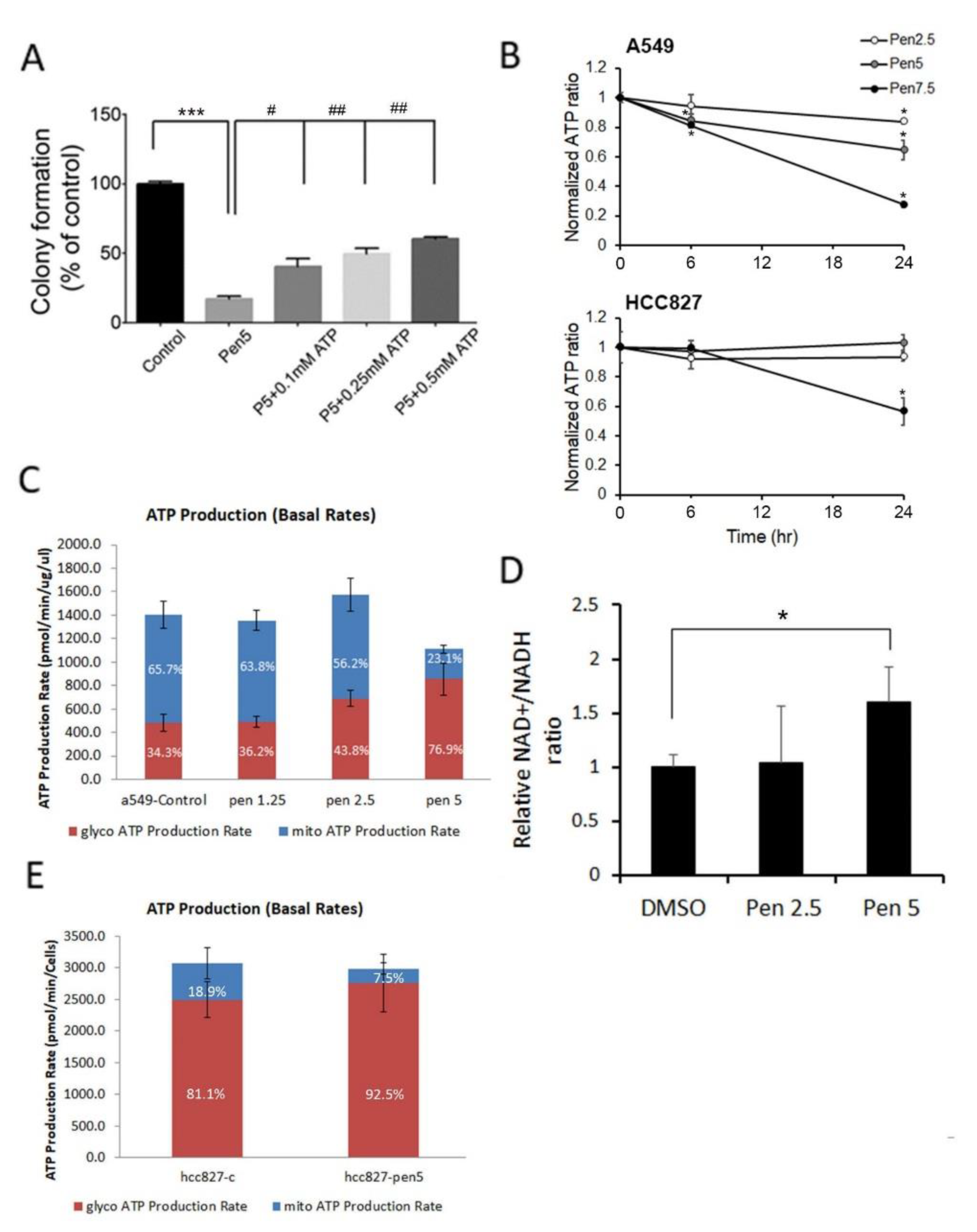
2.4. Intracellular ATP Detection Assay
2.5. ATP Production Rate of Glycolytic and Mitochondrial OXPHOS
2.6. NAD+ Measurement
2.7. Measurement of the Mitochondrial Mass
2.8. Total Cell Protein Lysate Extraction and Western Blot Analysis
2.9. Mitochondrial Membrane Potential (MMP) Assessment
2.10. Bioinformatics Analysis
2.11. Gene Expression Analysis by a Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR) Analysis
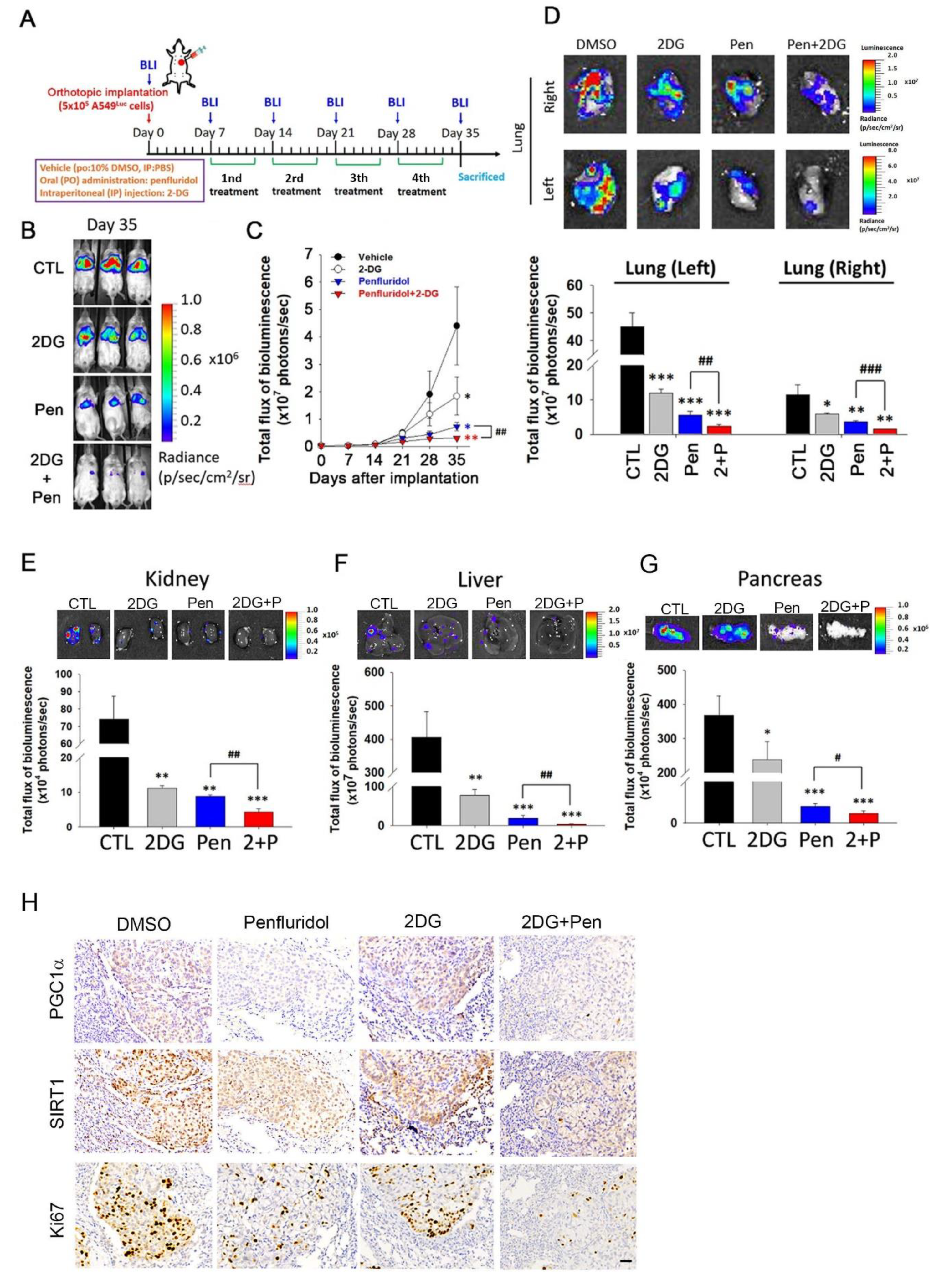
2.12. Lung Orthotopic Xenograft Tumor Model
2.13. Immunohistochemical (IHC) Staining
2.14. Statistical Analysis
3. Results
3.1. Penfluridol Treatment Alters Energy Metabolism in NSCLC Cells
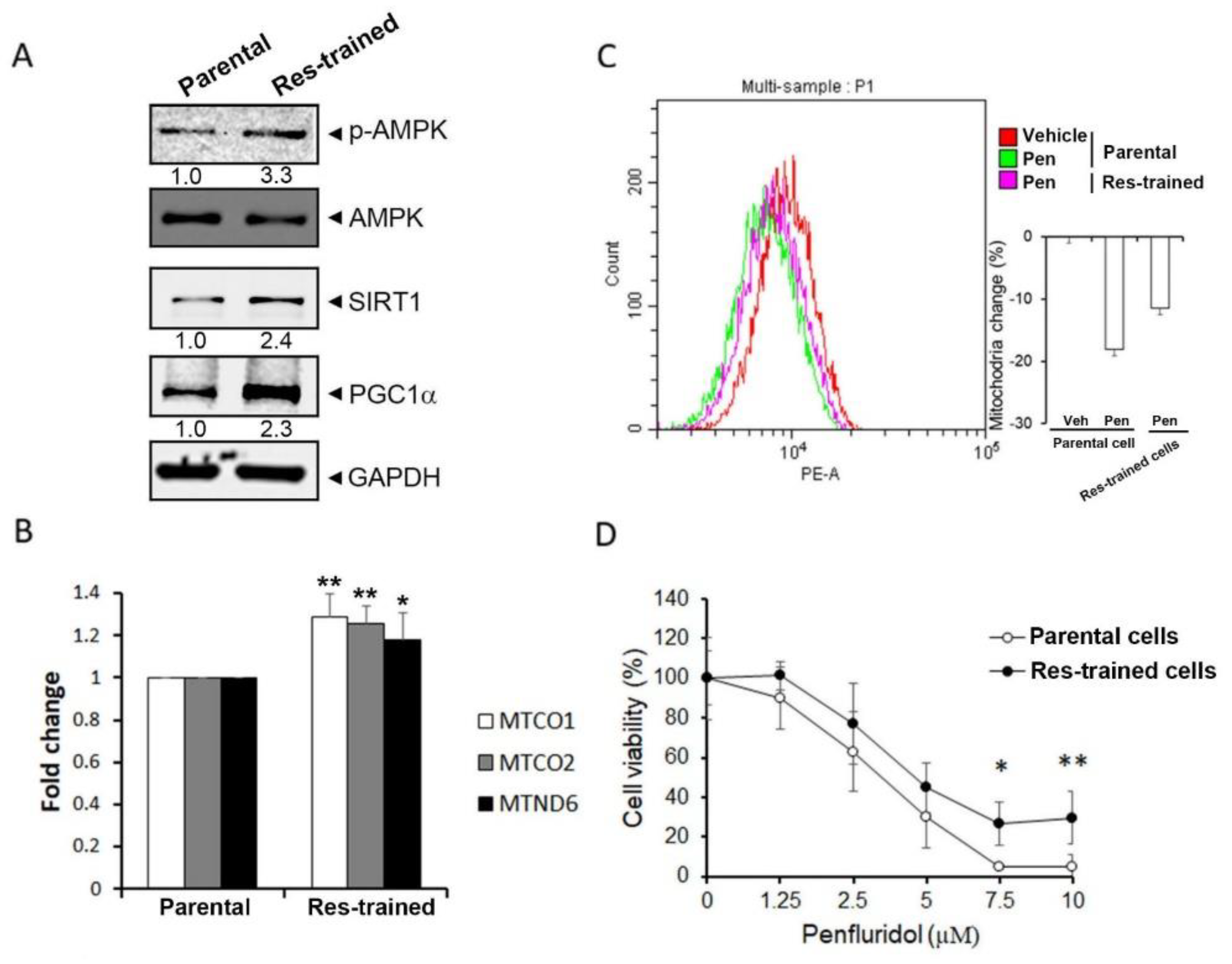
3.2. Penfluridol-Triggered Loss of Mitochondrial Function and Decrease in Mitochondrial Biogenesis via Downregulating PGC-1α in NSCLC Cells
3.3. Inhibition of Glycolysis by 2DG Promotes the Growth Inhibitory Effect of Penfluridol in NSCLC Cells
3.4. The SIRT1/PGC-1α Axis Is Critical for Penfluridol-Regulated Mitochondrial Biogenesis and Cell Viability of NSCLC Cells
3.5. Penfluridol in Combination with 2DG Dramatically Suppresses Tumor Progression in an A549 Orthotopic Graft Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar]
- Kryczka, J.; Kryczka, J.; Czarnecka-Chrebelska, K.H.; Brzeziańska-Lasota, E. Molecular Mechanisms of Chemoresistance Induced by Cisplatin in NSCLC Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 8885. [Google Scholar] [CrossRef] [PubMed]
- Morandi, A.; Indraccolo, S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 1–6. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The Role of PGC1α in Cancer Metabolism and its Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1α-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, S.; Li, J.; Zheng, L.; Feng, M.; Wang, X.; Han, K.; Pi, H.; Li, M.; Huang, X.; et al. SIRT1 facilitates hepatocellular carcinoma metastasis by promoting PGC-1α-mediated mitochondrial biogenesis. Oncotarget 2016, 7, 29255–29274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Xia, L.; Xiang, W.; He, W.; Yin, H.; Wang, F.; Gao, T.; Qi, W.; Yang, Z.; Yang, X.; et al. Metformin selectively inhibits metastatic colorectal cancer with the KRAS mutation by intracellular accumulation through silencing MATE. Proc. Natl. Acad. Sci. USA 2020, 117, 13012–13022. [Google Scholar] [CrossRef]
- Veschi, S.; De Lellis, L.; Florio, R.; Lanuti, P.; Massucci, A.; Tinari, N.; De Tursi, M.; di Sebastiano, P.; Marchisio, M.; Natoli, C.; et al. Effects of repurposed drug candidates nitroxoline and nelfinavir as single agents or in combination with erlotinib in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 236. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.H.; Tsai, K.Y.; Su, C.Y.; Lee, C.C. The incidence and relative risk factors for developing cancer among patients with schizophrenia: A nine-year follow-up study. Schizophr. Res. 2011, 129, 97–103. [Google Scholar] [CrossRef]
- Soares, B.G.; Lima, M.S. Penfluridol for schizophrenia. Cochrane Database Syst. Rev. 2006, 2, CD002923. [Google Scholar] [CrossRef]
- Shaw, V.; Srivastava, S.; Srivastava, S.K. Repurposing antipsychotics of the diphenylbutylpiperidine class for cancer therapy. Semin. Cancer Biol. 2021, 68, 75–83. [Google Scholar] [CrossRef]
- Tuan, N.M.; Lee, C.H. Penfluridol as a Candidate of Drug Repurposing for Anticancer Agent. Molecules 2019, 24, 3659. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, A.; Wright, S.; Srivastava, S.K. Immune consequences of penfluridol treatment associated with inhibition of glioblastoma tumor growth. Oncotarget 2017, 8, 47632–47641. [Google Scholar] [CrossRef] [Green Version]
- Tung, M.C.; Lin, Y.W.; Lee, W.J.; Wen, Y.C.; Liu, Y.C.; Chen, J.Q.; Hsiao, M.; Yang, Y.C.; Chien, M.H. Targeting DRD2 by the antipsychotic drug, penfluridol, retards growth of renal cell carcinoma via inducing stemness inhibition and autophagy-mediated apoptosis. Cell Death Dis. 2022, 13, 400. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, Y.Y.; Li, Z.X.; Zhao, Q.; Wang, X.; Yu, Y.; Wang, Y.Y.; Wang, Y.Q.; Luo, F. Anti-tumor effects of penfluridol through dysregulation of cholesterol homeostasis. Asian Pac. J. Cancer Prev. 2014, 15, 489–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, W.Y.; Chang, J.H.; Cheng, Y.; Cheng, G.Z.; Huang, H.C.; Hsiao, M.; Chung, C.L.; Lee, W.J.; Chien, M.H. Autophagosome accumulation-mediated ATP energy deprivation induced by penfluridol triggers nonapoptotic cell death of lung cancer via activating unfolded protein response. Cell. Death Dis. 2019, 10, 538. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, M.H.; Lee, W.J.; Yang, Y.C.; Tan, P.; Pan, K.F.; Liu, Y.C.; Tsai, H.C.; Hsu, C.H.; Wen, Y.C.; Hsiao, M.; et al. N-alpha-acetyltransferase 10 protein promotes metastasis by stabilizing matrix metalloproteinase-2 protein in human osteosarcomas. Cancer Lett. 2018, 433, 86–98. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef] [Green Version]
- Maynard, A.G.; Kanarek, N. NADH Ties One-Carbon Metabolism to Cellular Respiration. Cell Metab. 2020, 31, 660–662. [Google Scholar] [CrossRef]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio. Protoc. 2019, 9, e3128. [Google Scholar] [CrossRef]
- Taguchi, A.; Delgado, O.; Celiktaş, M.; Katayama, H.; Wang, H.; Gazdar, A.F.; Hanash, S.M. Proteomic signatures associated with p53 mutational status in lung adenocarcinoma. Proteomics 2014, 14, 2750–2759. [Google Scholar] [CrossRef] [Green Version]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Cannata, S.M.; Rotilio, G.; Ciriolo, M.R. p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid. Redox Signal. 2013, 18, 386–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, J.; Cao, N.; Li, Z.; Han, J.; Li, L. Resveratrol, an activator of SIRT1, induces protective autophagy in non-small-cell lung cancer via inhibiting Akt/mTOR and activating p38-MAPK. Onco. Targets Ther. 2018, 11, 7777–7786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lu, J.; Zhan, L.; Wang, M.; Shi, R.; Yuan, X.; Gao, X.; Liu, X.; Zang, J.; Liu, W.; et al. Resveratrol-induced Sirt1 phosphorylation by LKB1 mediates mitochondrial metabolism. J. Biol. Chem. 2021, 297, 100929. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, C.F.; Montal, E.D.; Haley, J.A.; Bott, A.J.; Haley, J.D. SREBP1 regulates mitochondrial metabolism in oncogenic KRAS expressing NSCLC. FASEB J. 2020, 34, 10574–10589. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Luo, X.; Ye, X.; Li, H.; Liu, H.; Du, Q.; Zhai, Q. SIRT1/PGC-1α/PPAR-γ Correlate with Hypoxia-Induced Chemoresistance in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 682762. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Li, Y.; Gu, S.; Chen, Y.; Yu, B.; Su, J.; Sun, L.; Liu, Y. p53 Affects PGC1α Stability Through AKT/GSK-3β to Enhance Cisplatin Sensitivity in Non-Small Cell Lung Cancer. Front. Oncol. 2020, 10, 1252. [Google Scholar] [CrossRef]
- Amat, R.; Planavila, A.; Chen, S.L.; Iglesias, R.; Giralt, M.; Villarroya, F. SIRT1 controls the transcription of the peroxisome proliferator-activated receptor-gamma Co-activator-1alpha (PGC-1alpha) gene in skeletal muscle through the PGC-1alpha autoregulatory loop and interaction with MyoD. J. Biol. Chem. 2009, 284, 21872–21880. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wang, Q.; Bai, L.; Chen, W.; Wang, X.; Tellez, C.S.; Leng, S.; Padilla, M.T.; Nyunoya, T.; Belinsky, S.A.; et al. RIP1 maintains DNA integrity and cell proliferation by regulating PGC-1α-mediated mitochondrial oxidative phosphorylation and glycolysis. Cell. Death Differ. 2014, 21, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007, 292, C125–C136. [Google Scholar] [CrossRef] [Green Version]
- Doménech, E.; Maestre, C.; Esteban-Martínez, L.; Partida, D.; Pascual, R.; Fernández-Miranda, G.; Seco, E.; Campos-Olivas, R.; Pérez, M.; Megias, D.; et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat. Cell Biol. 2015, 17, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Li, F.L.; Liu, J.P.; Bao, R.X.; Yan, G.; Feng, X.; Xu, Y.P.; Sun, Y.P.; Yan, W.; Ling, Z.Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, T.-C.; Lee, Y.-L.; Lee, W.-J.; Hung, W.-Y.; Cheng, G.-Z.; Chen, J.-Q.; Hsiao, M.; Chien, M.-H.; Chang, J.-H. Synergistic Tumor Inhibition via Energy Elimination by Repurposing Penfluridol and 2-Deoxy-D-Glucose in Lung Cancer. Cancers 2022, 14, 2750. https://doi.org/10.3390/cancers14112750
Lai T-C, Lee Y-L, Lee W-J, Hung W-Y, Cheng G-Z, Chen J-Q, Hsiao M, Chien M-H, Chang J-H. Synergistic Tumor Inhibition via Energy Elimination by Repurposing Penfluridol and 2-Deoxy-D-Glucose in Lung Cancer. Cancers. 2022; 14(11):2750. https://doi.org/10.3390/cancers14112750
Chicago/Turabian StyleLai, Tsung-Ching, Yueh-Lun Lee, Wei-Jiunn Lee, Wen-Yueh Hung, Guo-Zhou Cheng, Ji-Qing Chen, Michael Hsiao, Ming-Hsien Chien, and Jer-Hwa Chang. 2022. "Synergistic Tumor Inhibition via Energy Elimination by Repurposing Penfluridol and 2-Deoxy-D-Glucose in Lung Cancer" Cancers 14, no. 11: 2750. https://doi.org/10.3390/cancers14112750