
APOBEC Alteration Contributes to Tumor Growth and Immune Escape in Pan-Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome-Wide Omics Data and Clinical Data across 33 Cancer Types
2.2. Identification of Differentially Expressed Genes (DEGs)
2.3. Unsupervised Clustering for 11 APOBEC Genes
2.4. Estimation of TME Cell Infiltration
2.5. DEGs between Distinct Clusters and Functional Enrichment Analysis
2.6. Statistical Analysis
3. Results and Discussion
3.1. The Landscape of Genetic Alteration of APOBEC Family across Cancer Types
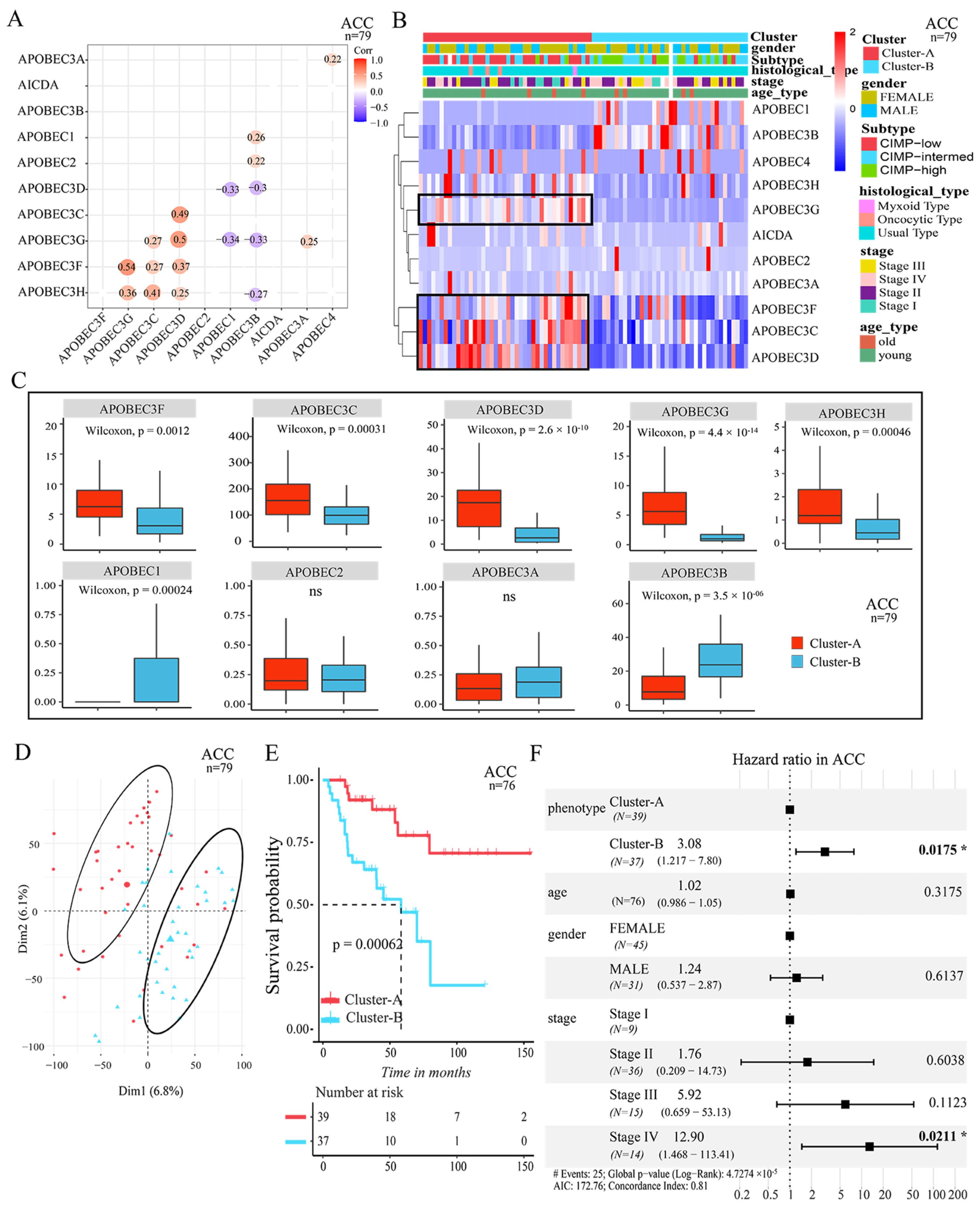
3.2. Two Patterns Mediated by APOBEC Family Were Significantly Correlated with Survival
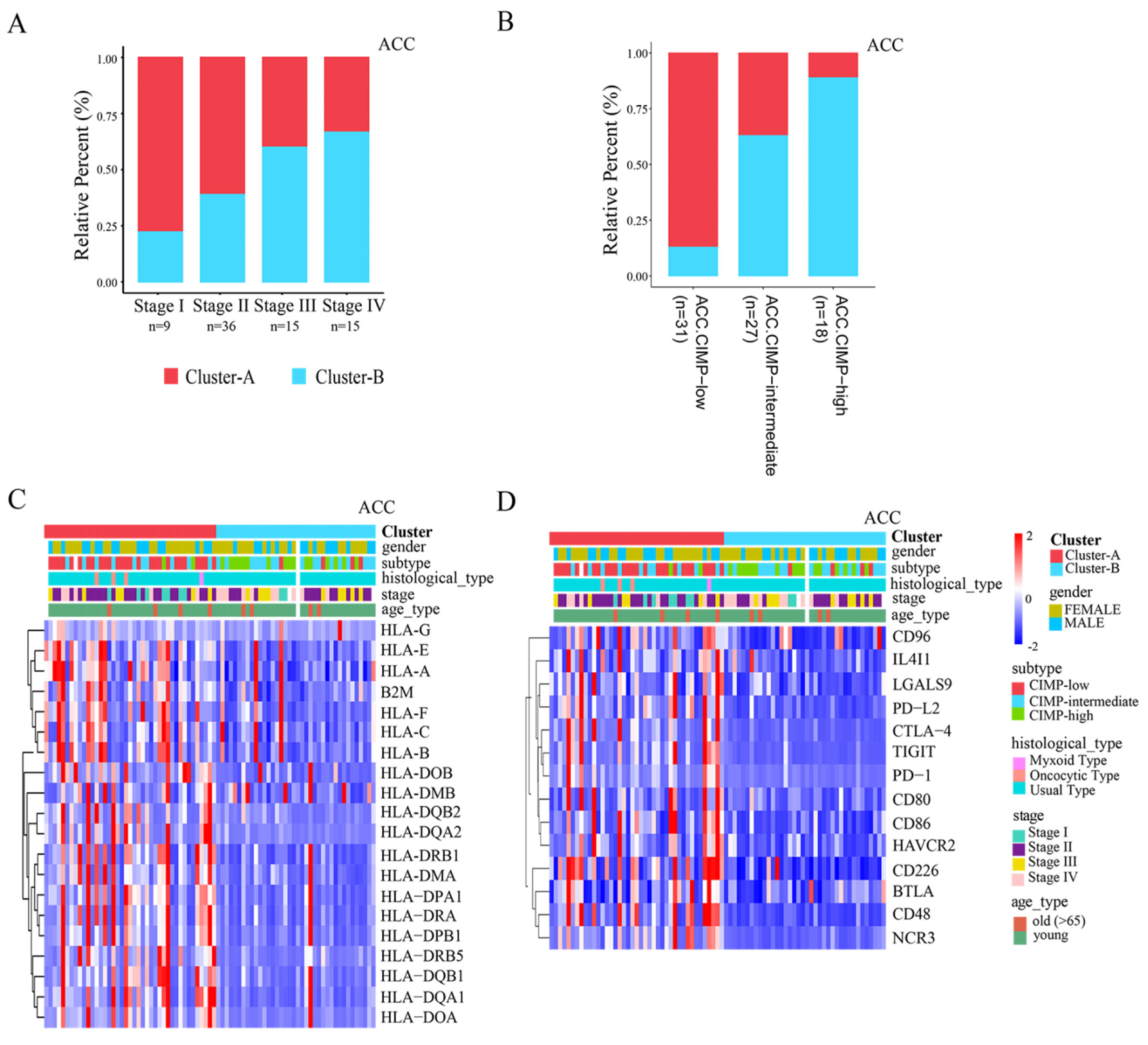
3.3. Distinct TME Infiltration Characteristics in the Two AMS Patterns
3.4. Clinical Relevance and Immunotherapy Sensitivity Association of AMS Pattern
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CIMP-high | CpG island hypermethylation |
| ICB | immunological checkpoint blockade |
| TME | tumor microenvironment |
| TSG | tumor suppressor gene |
| FPKM | fragments per kilobase per million |
| TPM | transcripts per kilobase million |
| AMS | APOBEC-mediated stratification |
| DEGs | differentially expressed genes |
| FDR | false discovery rate |
| GO | gene ontology |
| GSEA | gene set enrichment analysis |
| CNVs | copy number variations |
| HLAs | human leukocyte antigens |
References
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Revathidevi, S.; Murugan, A.K.; Nakaoka, H.; Inoue, I.; Munirajan, A.K. Apobec: A Molecular Driver in Cervical Cancer Pathogenesis. Cancer Lett. 2021, 496, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Akhter, J.; Aziz, M.A.A.; Al Ajlan, A.; Tulbah, A.; Akhtar, M. Breast Cancer: Is There A Viral Connection? Adv. Anat. Pathol. 2014, 21, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Fenton, T. Apobec3 Genes: Retroviral Restriction Factors to Cancer Drivers. Trends Mol. Med. 2015, 21, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.S. Molecular Mechanism and Clinical Impact of Apobec3b-Catalyzed Mutagenesis in Breast Cancer. Breast Cancer Res. 2015, 17, 8. [Google Scholar] [CrossRef] [Green Version]
- Moris, A.; Murray, S.; Cardinaud, S. Aid and Apobecs Span The Gap between Innate and Adaptive Immunity. Front. Microbiol. 2014, 5, 534. [Google Scholar] [CrossRef] [Green Version]
- Swanton, C.; McGranahan, N.; Starrett, G.J.; Harris, R.S. Apobec Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015, 5, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Kouno, T.; Silvas, T.V.; Hilbert, B.J.; Shandilya, S.; Bohn, M.F.; Kelch, B.A.; Royer, W.E.; Somasundaran, M.; Yilmaz, N.K.; Matsuo, H.; et al. Crystal Structure of Apobec3a Bound to Single-Stranded DNA Reveals Structural Basis for Cytidine Deamination and Specificity. Nat. Commun. 2017, 8, 15024. [Google Scholar] [CrossRef] [Green Version]
- Silvas, T.V.; Hou, S.; Myint, W.; Nalivaika, E.; Somasundaran, M.; Kelch, B.A.; Matsuo, H.; Yilmaz, N.K.; Schiffer, C.A. Substrate Sequence Selectivity of Apobec3a Implicates Intra-DNA Interactions. Sci. Rep. 2018, 8, 7511. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, S.J.; Liu, C.; Landau, M.; Buzovetsky, O.; Desimmie, B.A.; Zhao, Q.; Sasaki, T.; Burdick, R.C.; Pathak, V.K.; Anderson, K.S.; et al. Insights into DNA Substrate Selection by Apobec3g from Structural, Biochemical, and Functional Studies. PLoS ONE 2018, 13, e0195048. [Google Scholar] [CrossRef] [Green Version]
- Navaratnam, N.; Sarwar, R. An Overview of Cytidine Deaminases. Int. J. Hematol. 2006, 83, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An Apobec Cytidine Deaminase Mutagenesis Pattern Is Widespread in Human Cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for Apobec3b Mutagenesis in Multiple Human Cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caval, V.; Suspène, R.; Shapira, M.; Vartanian, J.P.; Wain-Hobson, S. A Prevalent Cancer Susceptibility Apobec3a Hybrid Allele Bearing Apobec3b 3’utr Enhances Chromosomal DNA Damage. Nat. Commun. 2014, 5, 5129. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Rosenthal, R.; Kanu, N.; McGranahan, N.; Bartek, J.; Quezada, S.A.; Hare, J.; Harris, R.S.; Swanton, C. Perspective: Apobec Mutagenesis in Drug Resistance and Immune Escape in HIV and Cancer Evolution. Ann. Oncol. 2018, 29, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. Cosmic: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Lever, J.; Zhao, E.Y.; Grewal, J.; Jones, M.R.; Jones, S.J. Cancermine: A Literature-Mined Resource for Drivers, Oncogenes and Tumor Suppressors in Cancer. Nat. Methods 2019, 16, 505–507. [Google Scholar] [CrossRef]
- Zhao, M.; Sun, J.; Zhao, Z. Tsgene: A Web Resource for Tumor Suppressor Genes. Nucleic Acids Res. 2013, 41, D970–D976. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting Neoantigens to Augment Antitumour Immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van EenooGhizlane Rouas, F.; Francis, P.; Crown, J.P.A.; Hitre, E.; de Azambuja, E.; et al. Prognostic and Predictive Value of Tumor-Infiltrating Lymphocytes in A Phase Iii Randomized Adjuvant Breast Cancer Trial in Node-Positive Breast Cancer Comparing The Addition of Docetaxel to Doxorubicin with Doxorubicin-Based Chemotherapy: Big 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, C.B.; Schuelke, M.R.; Kottke, T.; Thompson, J.M.; Wongthida, P.; Tonne, J.M.; Wongthida, P.; Tonne, J.M.; Huff, A.L.; Miller, A.; et al. Apobec3b-Mediated Corruption of the Tumor Cell Immunopeptidome Induces Heteroclitic Neoepitopes for Cancer Immunotherapy. Nat. Commun. 2020, 11, 790. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wen, W.; Bao, J.; Kuhs, K.L.; Cai, Q.; Long, J.; Shu, S.; Zheng, W.; Guo, X. Integrative Genomic Analyses of Apobec-Mutational Signature, Expression and Germline Deletion Of Apobec3 Genes, and Immunogenicity in Multiple Cancer Types. BMC Med. Genom. 2019, 12, 131. [Google Scholar] [CrossRef] [Green Version]
- DiMarco, A.V.; Qin, X.; McKinney, B.; Garcia, N.M.G.; Van Alsten, S.C.; Mendes, E.A.; Force, J.; Hanks, B.A.; Troester, M.A.; Owzar, K.; et al. Apobec Mutagenesis Inhibits Breast Cancer Growth Through Induction of T Cell-Mediated Antitumor Immune Responses. Cancer Immunol. Res. 2022, 10, 70–86. [Google Scholar] [CrossRef]
- Wang, S.; Jia, M.; He, Z.; Liu, X.S. Apobec3b and Apobec Mutational Signature as Potential Predictive Markers for Immunotherapy Response in Non-Small Cell Lung Cancer. Oncogene 2018, 37, 3924–3936. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Vokes, N.I.; Liu, D.; Taylor-Weiner, A.; Wankowicz, S.M.; Adeegbe, D.; Keliher, D.; Schilling, B.; Tracy, A.; et al. Genomic Correlates of Response to Immune Checkpoint Blockade In Microsatellite-Stable Solid Tumors. Nat. Genet. 2018, 50, 1271–1278. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Jain, E.; Cohen, O.; Kim, D.; Buendia-Buendia, J.; Winer, E.; Lin, N.; Tolaney, S.M.; Wagle, N. Prevalence and Mutational Determinants of High Tumor Mutation Burden in Breast Cancer. Ann. Oncol. 2020, 31, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. Tcgabiolinks: An R/Bioconductor Package for Integrative Analysis of Tcga Data. Nucleic Acids Res. 2016, 44, E71. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. Gistic2.0 Facilitates Sensitive and Confident Localization of the Targets of Focal Somatic Copy-Number Alteration in Human Cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [Green Version]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved Pan-Cancer Microenvironment Subtypes Predict Response to Immunotherapy. Cancer Cell 2021, 39, 845–865.e7. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for Rna-Seq Data with Deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkerson, M.D.; Hayes, D.N. Consensusclusterplus: A Class Discovery Tool with Confidence Assessments and Item Tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring Tumour Purity and Stromal and Immune Cell Admixture from Expression Data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust Enumeration of Cell Subsets from Tissue Expression Profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. Clusterprofiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Hazra, A.; Gogtay, N. Biostatistics Series Module 3: Comparing Groups: Numerical Variables. Indian J. Dermatol. 2016, 61, 251–260. [Google Scholar] [CrossRef]
- Sebestyén, E.; Singh, B.; Miñana, B.; Pagès, A.; Mateo, F.; Pujana, M.A.; Valcárcel, J.; Eyras, E. Large-Scale Analysis of Genome and Transcriptome Alterations in Multiple Tumors Unveils Novel Cancer-Relevant Splicing Networks. Genome Res. 2016, 26, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 175, 598–599. [Google Scholar] [CrossRef] [Green Version]
- Perri, F.; Longo, F.; Giuliano, M.; Sabbatino, F.; Favia, G.; Ionna, F.; Addeo, R.; Della Vittoria Scarpati, G.; di Lorenzo, G.; Pisconti, S. Epigenetic Control of Gene Expression: Potential Implications for Cancer Treatment. Crit. Rev. Oncol. Hematol. 2017, 111, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Else, T.; Williams, A.R.; Sabolch, A.; Jolly, S.; Miller, B.S.; Hammer, G.D. Adjuvant Therapies and Patient and Tumor Characteristics Associated with Survival of Adult Patients with Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2014, 99, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassnacht, M.; Kroiss, M.; Allolio, B. Update in Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, 4551–4564. [Google Scholar] [CrossRef] [PubMed]
- Barreau, O.; Assie, G.; Wilmot-Roussel, H.; Ragazzon, B.; Baudry, C.; Perlemoine, K.; René-Corail, F.; Bertagna, X.; Dousset, B.; Hamzaoui, N.; et al. Identification of A Cpg Island Methylator Phenotype in Adrenocortical Carcinomas. J. Clin. Endocrinol. Metab. 2013, 98, E174–E184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive Genomic Profiles of Small Cell Lung Cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Zhu, L.; Huang, L.; Sun, Z.; Zhang, H.; Nong, B.; Xiong, Y. APOBEC Alteration Contributes to Tumor Growth and Immune Escape in Pan-Cancer. Cancers 2022, 14, 2827. https://doi.org/10.3390/cancers14122827
Guo H, Zhu L, Huang L, Sun Z, Zhang H, Nong B, Xiong Y. APOBEC Alteration Contributes to Tumor Growth and Immune Escape in Pan-Cancer. Cancers. 2022; 14(12):2827. https://doi.org/10.3390/cancers14122827
Chicago/Turabian StyleGuo, Honghong, Ling Zhu, Lu Huang, Zhen Sun, Hui Zhang, Baoting Nong, and Yuanyan Xiong. 2022. "APOBEC Alteration Contributes to Tumor Growth and Immune Escape in Pan-Cancer" Cancers 14, no. 12: 2827. https://doi.org/10.3390/cancers14122827