
Spinal Solitary Fibrous Tumors: An Original Multicenter Series and Systematic Review of Presentation, Management, and Prognosis

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Original Series
2.2. Systematic Review
2.3. Statistical Analysis
3. Results
3.1. Patients
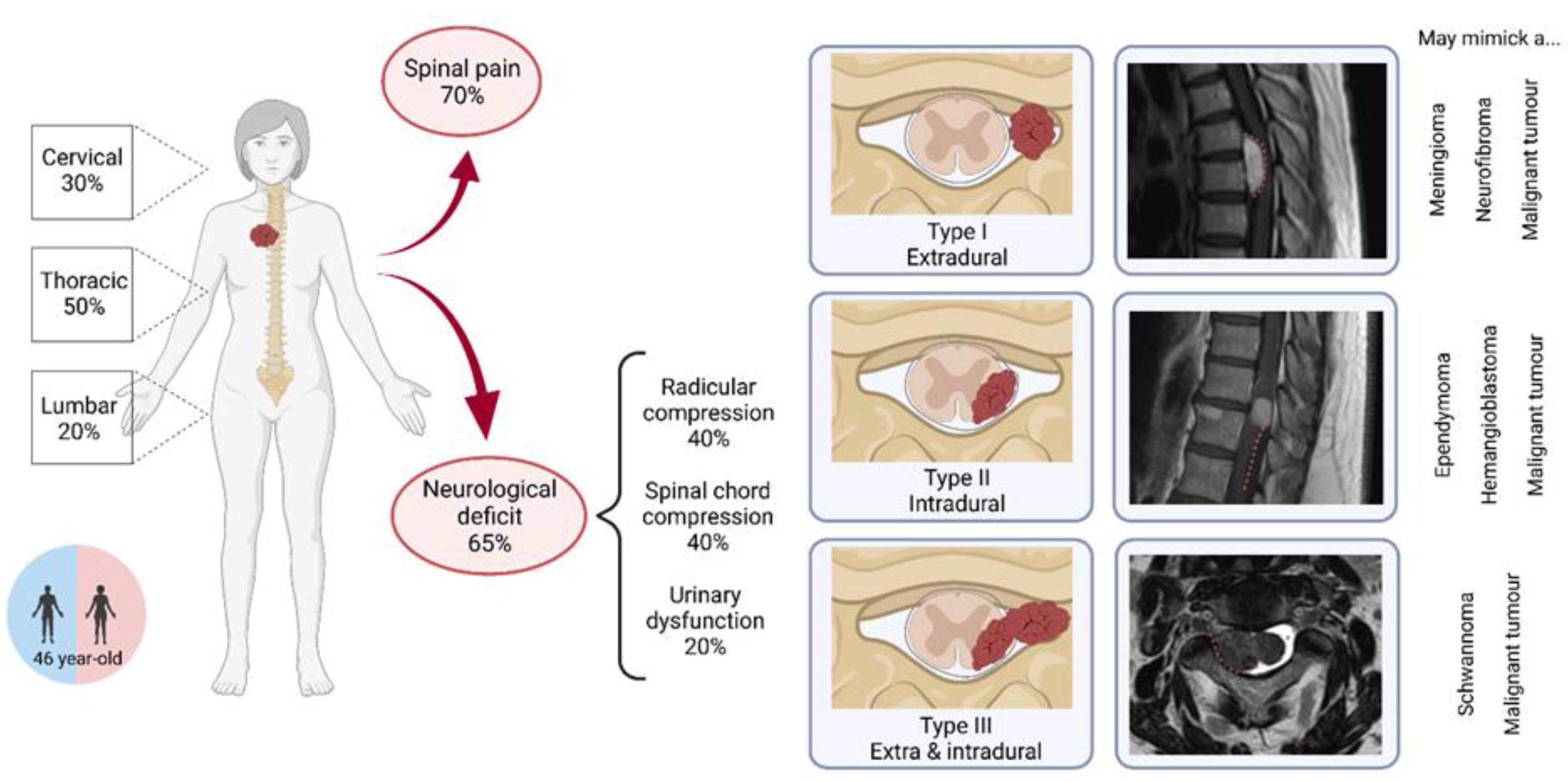
3.2. Clinical Presentation
3.3. Initial Radiological Findings
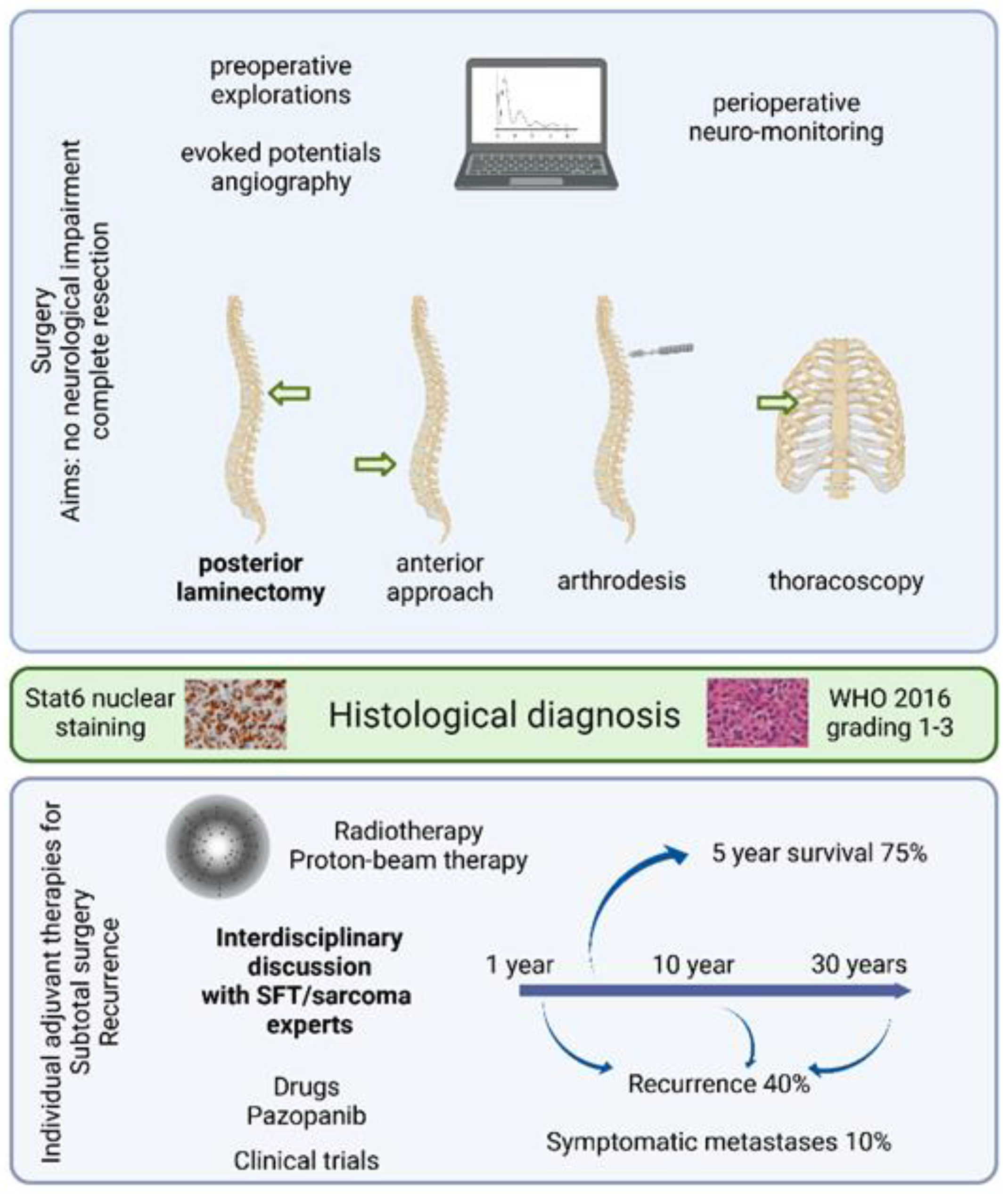
3.4. Operative Findings
3.5. Histological Findings
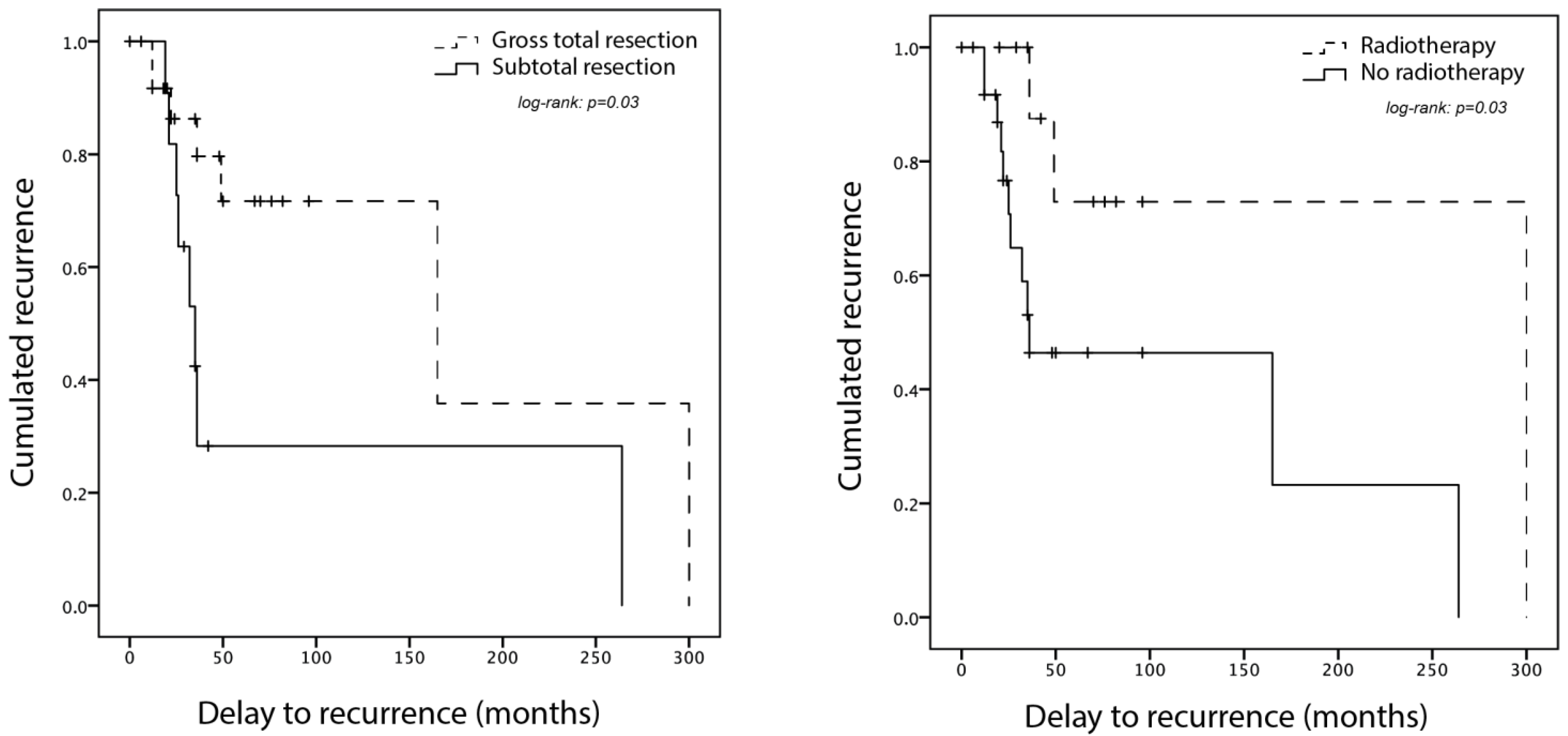
3.6. Adjuvant Treatment and Outcome
4. Discussion
4.1. Clinical Approach to Spinal SFT Management Based on Data and Experience
4.2. Postoperative Decision Making in Spinal SFT Treatment
4.3. Specificities of Spinal SFT
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.-M.; Kalyana-Sundaram, S.; Cao, X.; Lonigro, R.J.; Sung, Y.-S.; Chen, C.-L.; Zhang, L.; Wang, R.; Su, F.; et al. Identification of Recurrent NAB2-STAT6 Gene Fusions in Solitary Fibrous Tumor by Integrative Sequencing. Nat. Genet. 2013, 45, 180–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Yang, A.; Chen, N.; Yang, J.; Qiu, X.; Zhang, J. Hemangiopericytomas in the Spine: Clinical Features, Classification, Treatment, and Long-Term Follow-up in 26 Patients. Neurosurgery 2013, 72, 16–24; discussion 24 . [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Ando, M.; Kobayashi, H.; Shinozaki-Ushiku, A.; Chikuda, H.; Matsubayashi, Y.; Yoshida, M.; Saito, Y.; Kohsaka, S.; Oda, K.; Miyagawa, K.; et al. Spinal Solitary Fibrous Tumor of the Neck: Next-Generation Sequencing-Based Analysis of Genomic Aberrations. Auris Nasus Larynx 2020, 47, 1058–1063. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, K.; Han, L.; Jiao, L.; Liu, W.; Xu, Y.; Niu, H.; Ke, C.; Shu, K.; Lei, T. Solitary Fibrous Tumor/Hemangiopericytoma of Spinal Cord: A Retrospective Single-Center Study of 16 Cases. World Neurosurg. 2019, 123, e629–e638. [Google Scholar] [CrossRef]
- Wang, L.; Yu, J.; Shu, D.; Huang, B.; Wang, Y.; Zhang, L. Primary Endodermal Hemangiopericytoma/Solitary Fibrous Tumor of the Cervical Spine: A Case Report and Literature Review. BMC Surg. 2021, 21, 405. [Google Scholar] [CrossRef]
- Su, H.-Y.; Tsai, T.-H.; Yang, S.-F.; Lee, J.-Y. Dumbbell-Shaped Solitary Fibrous Tumor of Thoracic Spine. Kaohsiung J. Med. Sci. 2019, 35, 517–518. [Google Scholar] [CrossRef]
- Flores-Justa, A.; López-García, E.; García-Allut, A.; Reyes-Santías, R.M. Solitary Fibrous Tumour/Haemangiopericytoma of the Spinal Cord. Neurocirugia (Astur. Engl. E) 2018, 29, 309–313. [Google Scholar] [CrossRef]
- Mansilla Fernández, B.; Román de Aragón, M.; Paz Solís, J.F.; García Feijoo, P.; Roda Frade, J.; Regojo Zapata, M.R. Solitary Fibrous Tumor: A Clinical Case. Neurocirugia (Astur. Engl. Ed.) 2019, 30, 33–37. [Google Scholar] [CrossRef]
- Olmsted, Z.T.; Tabor, J.; Doron, O.; Hosseini, H.; Schneider, D.; Green, R.; Wahl, S.J.; Sciubba, D.M.; D’Amico, R.S. Intradural Extramedullary Solitary Fibrous Tumor of the Thoracic Spinal Cord. Cureus 2021, 13, e18613. [Google Scholar] [CrossRef] [PubMed]
- Albert, G.W.; Gokden, M. Solitary Fibrous Tumors of the Spine: A Pediatric Case Report with a Comprehensive Review of the Literature. J. Neurosurg. Pediatr. 2017, 19, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauleac, C.; Vasiljevic, A.; Berhouma, M. How to Differentiate Spinal Cord Hemangiopericytoma from Common Spinal Cord Tumor? Neurochirurgie 2020, 66, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Endo, K.; Aihara, T.; Matsuoka, Y.; Nishimura, H.; Suzuki, H.; Sawaji, Y.; Yamamoto, K.; Fukami, S.; Tanigawa, M.; et al. Salvage Carbon Ion Radiotherapy for Recurrent Solitary Fibrous Tumor: A Case Report and Literature Review. J. Orthop. Surg. (Hong Kong) 2020, 28, 2309499019896099. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Ma, M.; Li, H. Invasive Solitary Fibrous Tumor/Hemangiopericytoma of the Filum Terminale. World Neurosurg. 2020, 139, 318–321. [Google Scholar] [CrossRef]
- Shukla, P.; Gulwani, H.V.; Kaur, S.; Shanmugasundaram, D. Reappraisal of Morphological and Immunohistochemical Spectrum of Intracranial and Spinal Solitary Fibrous Tumors/Hemangiopericytomas with Impact on Long-Term Follow-Up. Indian J. Cancer 2018, 55, 214–221. [Google Scholar] [CrossRef]
- Yao, Z.-G.; Wu, H.-B.; Hao, Y.-H.; Wang, X.-F.; Ma, G.-Z.; Li, J.; Li, J.-F.; Lin, C.-H.; Zhong, X.-M.; Wang, Z.; et al. Papillary Solitary Fibrous Tumor/Hemangiopericytoma: An Uncommon Morphological Form With NAB2-STAT6 Gene Fusion. J. Neuropathol. Exp. Neurol. 2019, 78, 685–693. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Xiao, Q.; Zeng, J.-H.; Deng, L. Solitary Fibrous Tumor of the Lumbar Spine Resembling Schwannoma: A Case Report and Review of the Literature. World Neurosurg. 2019, 124, 121–124. [Google Scholar] [CrossRef]
- Oike, N.; Kawashima, H.; Ogose, A.; Hotta, T.; Hirano, T.; Ariizumi, T.; Yamagishi, T.; Umezu, H.; Inagawa, S.; Endo, N. A Malignant Solitary Fibrous Tumour Arising from the First Lumbar Vertebra and Mimicking an Osteosarcoma: A Case Report. World J. Surg. Oncol. 2017, 15, 100. [Google Scholar] [CrossRef]
- Farooq, Z.; Badar, Z.; Zaccarini, D.; Tavernier, F.B.; Mohamed, A.; Mangla, R. Recurrent Solitary Fibrous Tumor of Lumbar Spine with Vertebral Body Involvement: Imaging Features and Differential Diagnosis with Report of a Case. Radiol. Case Rep. 2016, 11, 450–455. [Google Scholar] [CrossRef] [Green Version]
- Mariniello, G.; Napoli, M.; Russo, C.; Briganti, F.; Giamundo, A.; Maiuri, F.; Del Basso De Caro, M.L. MRI Features of Spinal Solitary Fibrous Tumors. A Report of Two Cases and Literature Review. Neuroradiol. J. 2012, 25, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Alleyne, C.H.; Cawley, C.M.; Shengelaia, G.G.; Barrow, D.L. Microsurgical Anatomy of the Artery of Adamkiewicz and Its Segmental Artery. Neurosurg. Focus 1998, 5, E2. [Google Scholar] [CrossRef] [Green Version]
- Santillan, A.; Zink, W.; Lavi, E.; Boockvar, J.; Gobin, Y.P.; Patsalides, A. Endovascular Embolization of Cervical Hemangiopericytoma with Onyx-18: Case Report and Review of the Literature. J. Neurointerv. Surg. 2011, 3, 304–307. [Google Scholar] [CrossRef] [PubMed]
- El Hindy, N.; Ringelstein, A.; Forsting, M.; Sure, U.; Mueller, O. Spinal Metastasis from Malignant Meningeal Intracranial Hemangiopericytoma: One-Staged Percutaneous OnyxTM Embolization and Resection—A Technical Innovation. World J. Surg. Oncol. 2013, 11, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millesi, M.; Kiesel, B.; Woehrer, A.; Hainfellner, J.A.; Novak, K.; Martínez-Moreno, M.; Wolfsberger, S.; Knosp, E.; Widhalm, G. Analysis of 5-Aminolevulinic Acid-Induced Fluorescence in 55 Different Spinal Tumors. Neurosurg. Focus 2014, 36, E11. [Google Scholar] [CrossRef] [PubMed]
- Apra, C.; Mokhtari, K.; Cornu, P.; Peyre, M.; Kalamarides, M. Intracranial Solitary Fibrous Tumors/Hemangiopericytomas: First Report of Malignant Progression. J. Neurosurg. 2018, 128, 1719–1724. [Google Scholar] [CrossRef] [Green Version]
- Demicco, E.G.; Wagner, M.J.; Maki, R.G.; Gupta, V.; Iofin, I.; Lazar, A.J.; Wang, W.-L. Risk Assessment in Solitary Fibrous Tumors: Validation and Refinement of a Risk Stratification Model. Mod. Pathol. 2017, 30, 1433–1442. [Google Scholar] [CrossRef]
- Apra, C.; Guillemot, D.; Frouin, E.; Bouvier, C.; Mokhtari, K.; Kalamarides, M.; Pierron, G. Molecular Description of Meningeal Solitary Fibrous Tumors/Hemangiopericytomas Compared to Meningiomas: Two Completely Separate Entities. J. Neurooncol. 2021, 154, 327–334. [Google Scholar] [CrossRef]
- Nakada, S.; Minato, H.; Nojima, T. Clinicopathological Differences between Variants of the NAB2-STAT6 Fusion Gene in Solitary Fibrous Tumors of the Meninges and Extra-Central Nervous System. Brain Tumor Pathol. 2016, 33, 169–174. [Google Scholar] [CrossRef]
- Reisenauer, J.S.; Mneimneh, W.; Jenkins, S.; Mansfield, A.S.; Aubry, M.C.; Fritchie, K.J.; Allen, M.S.; Blackmon, S.H.; Cassivi, S.D.; Nichols, F.C.; et al. Comparison of Risk Stratification Models to Predict Recurrence and Survival in Pleuropulmonary Solitary Fibrous Tumor. J. Thorac. Oncol. 2018, 13, 1349–1362. [Google Scholar] [CrossRef] [Green Version]
- Barthelmeß, S.; Geddert, H.; Boltze, C.; Moskalev, E.A.; Bieg, M.; Sirbu, H.; Brors, B.; Wiemann, S.; Hartmann, A.; Agaimy, A.; et al. Solitary Fibrous Tumors/Hemangiopericytomas with Different Variants of the NAB2-STAT6 Gene Fusion Are Characterized by Specific Histomorphology and Distinct Clinicopathological Features. Am. J. Pathol. 2014, 184, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.L.; Walraven, I.; Lecointe-Artzner, E.; van Houdt, W.J.; Scholten, A.N.; Strauss, D.; Schrage, Y.; Hayes, A.J.; Raut, C.P.; Fairweather, M.; et al. Management of Meningeal Solitary Fibrous Tumors/Hemangiopericytoma; Surgery Alone or Surgery plus Postoperative Radiotherapy? Acta Oncol. 2021, 60, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.L.; Walraven, I.; Lecointe-Artzner, E.; van Houdt, W.J.; Strauss, D.; Schrage, Y.; Hayes, A.J.; Raut, C.P.; Fairweather, M.; Baldini, E.H.; et al. Extrameningeal Solitary Fibrous Tumors-Surgery Alone or Surgery plus Perioperative Radiotherapy: A Retrospective Study from the Global Solitary Fibrous Tumor Initiative in Collaboration with the Sarcoma Patients EuroNet. Cancer 2020, 126, 3002–3012. [Google Scholar] [CrossRef] [PubMed]
- Dufour, H.; Métellus, P.; Fuentes, S.; Murracciole, X.; Régis, J.; Figarella-Branger, D.; Grisoli, F. Meningeal Hemangiopericytoma: A Retrospective Study of 21 Patients with Special Review of Postoperative External Radiotherapy. Neurosurgery 2001, 48, 756–762; discussion 762–763 . [Google Scholar]
- Apra, C.; Alentorn, A.; Mokhtari, K.; Kalamarides, M.; Sanson, M. Pazopanib Efficacy in Recurrent Central Nervous System Hemangiopericytomas. J. Neurooncol. 2018, 139, 369–372. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Tortoreto, M.; Baldi, G.G.; Grignani, G.; Toss, A.; Badalamenti, G.; Cominetti, D.; Morosi, C.; Dei Tos, A.P.; Festinese, F.; et al. Preclinical and Clinical Evidence of Activity of Pazopanib in Solitary Fibrous Tumour. Eur. J. Cancer 2014, 50, 3021–3028. [Google Scholar] [CrossRef]
- de Bernardi, A.; Dufresne, A.; Mishellany, F.; Blay, J.-Y.; Ray-Coquard, I.; Brahmi, M. Novel Therapeutic Options for Solitary Fibrous Tumor: Antiangiogenic Therapy and Beyond. Cancers 2022, 14, 1064. [Google Scholar] [CrossRef]
- Kawashima, M.; Suzuki, S.O.; Yamashima, T.; Fukui, M.; Iwaki, T. Prostaglandin D Synthase (Beta-Trace) in Meningeal Hemangiopericytoma. Mod. Pathol. 2001, 14, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Peyre, M.; Salaud, C.; Clermont-Taranchon, E.; Niwa-Kawakita, M.; Goutagny, S.; Mawrin, C.; Giovannini, M.; Kalamarides, M. PDGF Activation in PGDS-Positive Arachnoid Cells Induces Meningioma Formation in Mice Promoting Tumor Progression in Combination with Nf2 and Cdkn2ab Loss. Oncotarget 2015, 6, 32713–32722. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Characteristics | Our Series (n = 10) | Literature Review (n = 31) | Total (n = 41) | |
|---|---|---|---|---|
| Population | ||||
| Sex | M | 6 (60%) | 14 (45%) | 20 (49%) |
| F | 4 (40%) | 17 (55%) | 21 (51%) | |
| Age, mean ± IC95 | 47 ± 8 | 45 ± 3 | 46 ± 4 | |
| Clinical and radiological presentations | ||||
| Spinal pain | 5 (50%) | 22 (76%) | 27 (69%) | |
| Radicular compression | 6 (60%) | 8 (28%) | 14 (36%) | |
| Spinal cord compression | 7 (70%) | 9 (31%) | 16 (41%) | |
| Urinary dysfunction | 4(40%) | 3 (10%) | 7 (18%) | |
| Motor dysfunction | 5 (50%) | 9 (29%) | 14 (34%) | |
| Sensory dysfunction | 4 (40%) | 6 (19%) | 10 (24%) | |
| Duration of symptoms (mo) | 10 ± 6 | 20 ± 12 | 17 ± 9 | |
| Tumor localization | Cervical | 5 (50%) | 7 (23%) | 12 (29%) |
| Thoracic | 4 (40%) | 17 (54%) | 21 (51%) | |
| Lumbar | 1 (10%) | 7 (23%) | 8 (20%) | |
| Tumor type | I extradural | 0 (0%) | 2 (14%) | 2 (9%) |
| II intradural | 7 (78%) | 8 (57%) | 15 (65%) | |
| III extra- and intradural | 2 (22%) | 4 (29%) | 6 (26%) | |
| Surgical and histological findings | ||||
| Complete resection | 7 (70%) | 22 (71%) | 29 (71%) | |
| Purely extramedullary tumor during surgery | 5 (50%) | 19 (68%) | 24 (63%) | |
| Histological grading | 1 | 1 (11%) | 6 (55%) | 7 (35%) |
| 2 | 5 (56%) | 0 (0%) | 5 (25%) | |
| 3 | 3 (33%) | 5 (45%) | 8 (40%) | |
| Post-operative management and outcome | ||||
| Primary adjuvant treatment | None | 6 (60%) | 14 (77%) | 30 (73%) |
| Radiotherapy | 4 (40%) | 7 (23%) | 11 (27%) | |
| Documented recurrence | 4 (40%) | 11 (32%) | 15 (37%) | |
| Time to first recurrence (mo) | 128 ± 116 | 49 ± 42 | 70 ± 47 | |
| Risk Factor for Recurrence | Recurrence (n = 15) | No Recurrence (n = 20) | p-Value |
|---|---|---|---|
| Intramedullary component | 36% (n = 14) | 26% (n = 29) | 0.70 |
| Subtotal resection | 53% (n = 15) | 20% (n = 20) | 0.07 |
| WHO grade 3 | 40% (n = 5) | 50% (n = 10) | 1 |
| Adjuvant radiotherapy | 13% (n = 15) | 40% (n = 20) | 0.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apra, C.; El Arbi, A.; Montero, A.-S.; Parker, F.; Knafo, S. Spinal Solitary Fibrous Tumors: An Original Multicenter Series and Systematic Review of Presentation, Management, and Prognosis. Cancers 2022, 14, 2839. https://doi.org/10.3390/cancers14122839
Apra C, El Arbi A, Montero A-S, Parker F, Knafo S. Spinal Solitary Fibrous Tumors: An Original Multicenter Series and Systematic Review of Presentation, Management, and Prognosis. Cancers. 2022; 14(12):2839. https://doi.org/10.3390/cancers14122839
Chicago/Turabian StyleApra, Caroline, Amira El Arbi, Anne-Sophie Montero, Fabrice Parker, and Steven Knafo. 2022. "Spinal Solitary Fibrous Tumors: An Original Multicenter Series and Systematic Review of Presentation, Management, and Prognosis" Cancers 14, no. 12: 2839. https://doi.org/10.3390/cancers14122839
APA StyleApra, C., El Arbi, A., Montero, A.-S., Parker, F., & Knafo, S. (2022). Spinal Solitary Fibrous Tumors: An Original Multicenter Series and Systematic Review of Presentation, Management, and Prognosis. Cancers, 14(12), 2839. https://doi.org/10.3390/cancers14122839