
IFI35 Promotes Renal Cancer Progression by Inhibiting pSTAT1/pSTAT6-Dependent Autophagy


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Specimens and Ethics Statement
2.2. Immunohistochemistry (IHC) Staining
2.3. Western Blotting
2.4. Cell Culture and Stable Cell Line Selection
2.5. Cell Proliferation Assay
2.6. Cell Apoptosis Analysis
2.7. Cell Invasion Assay
2.8. Analysis of Autophagy by Fluorescence Microscope
2.9. Flow Cytometry Analysis
2.10. Animal Models
2.11. Pathological Analysis
2.12. Statistical Analysis
3. Results
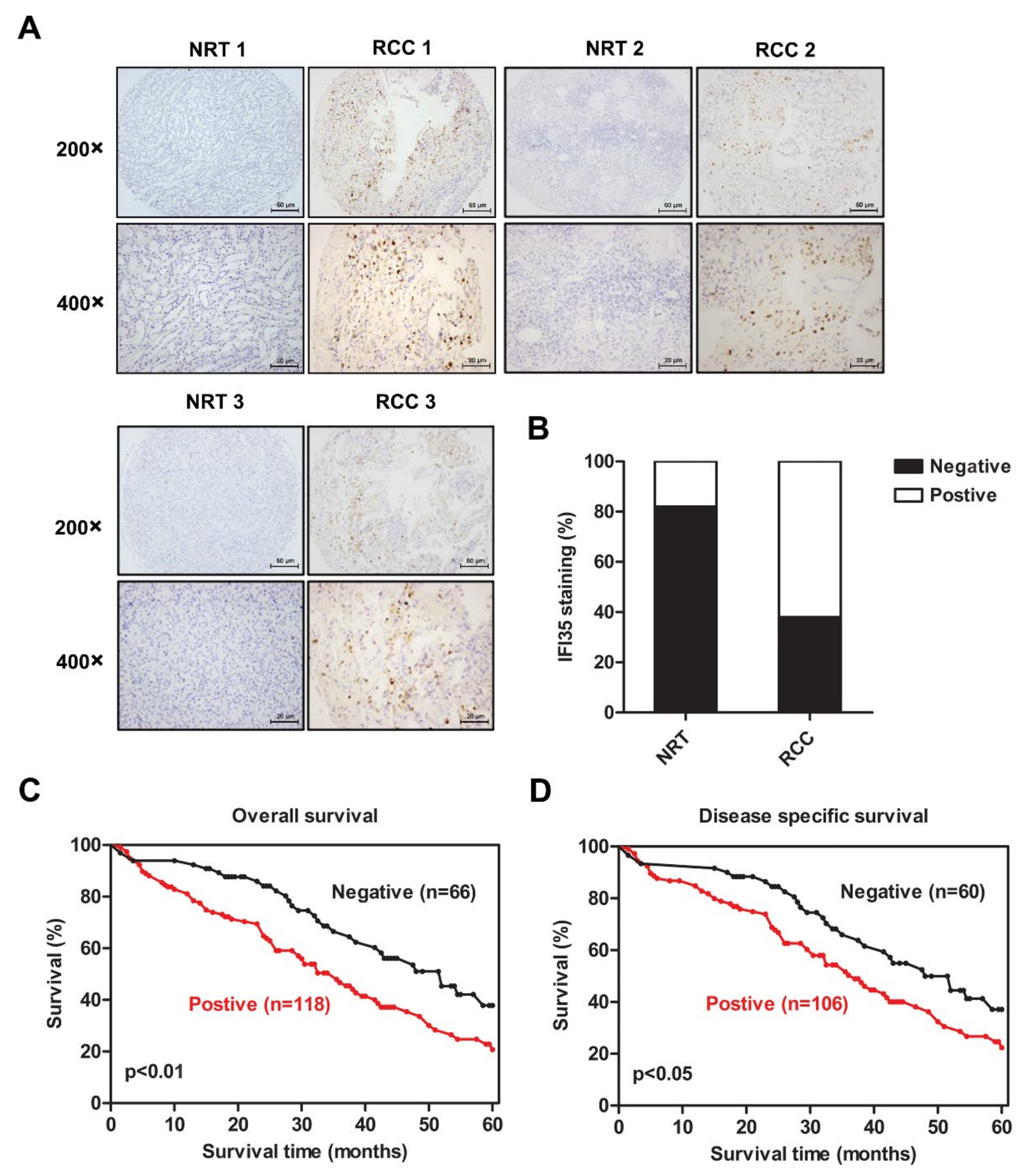
3.1. Increased Expression of IFI35 Is Correlated with Poor Prognosis in RCC
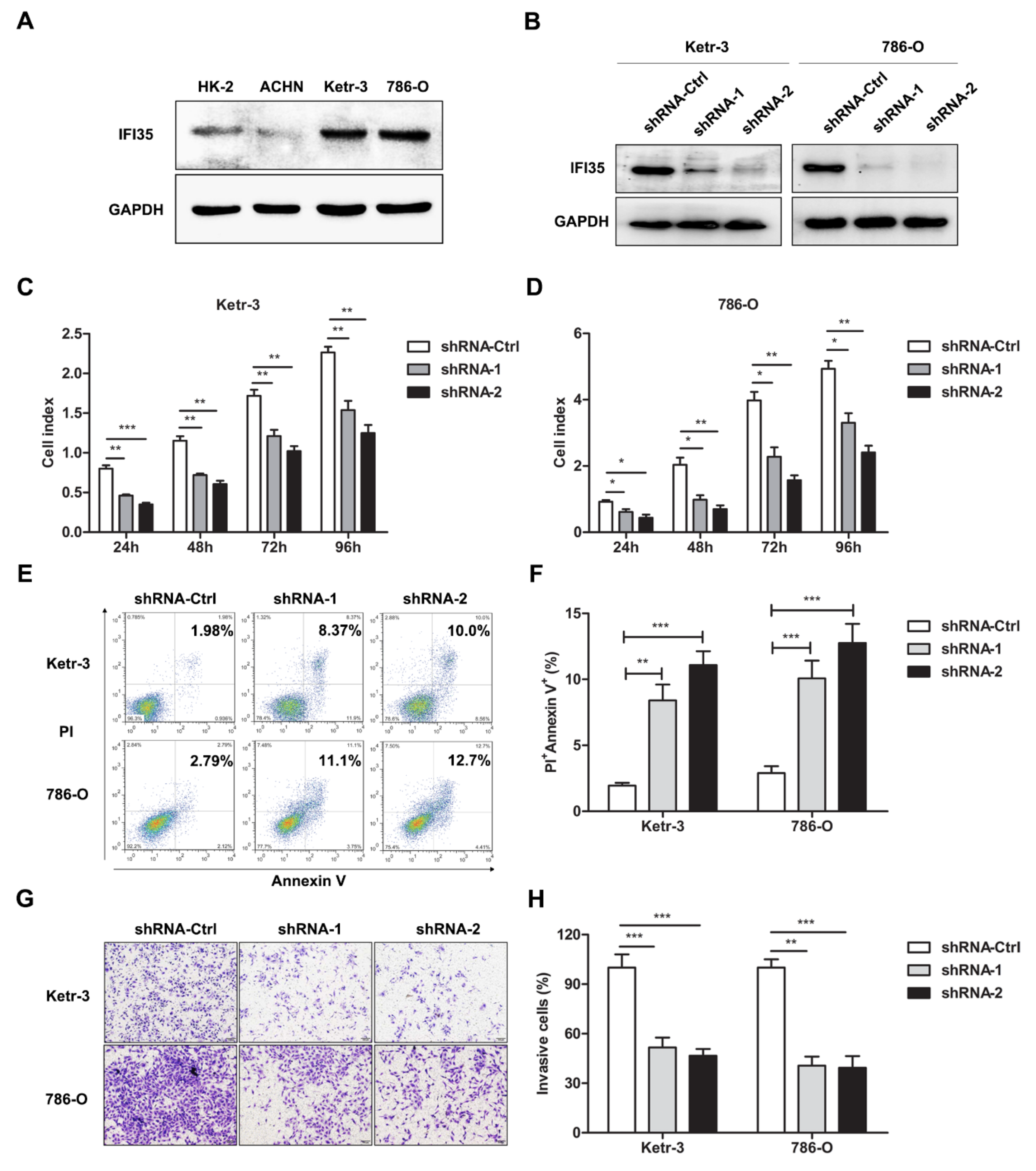
3.2. Knockdown of IFI35 Inhibits Cell Proliferation and Invasion and Promotes Cell Apoptosis in Renal Cancer Cells
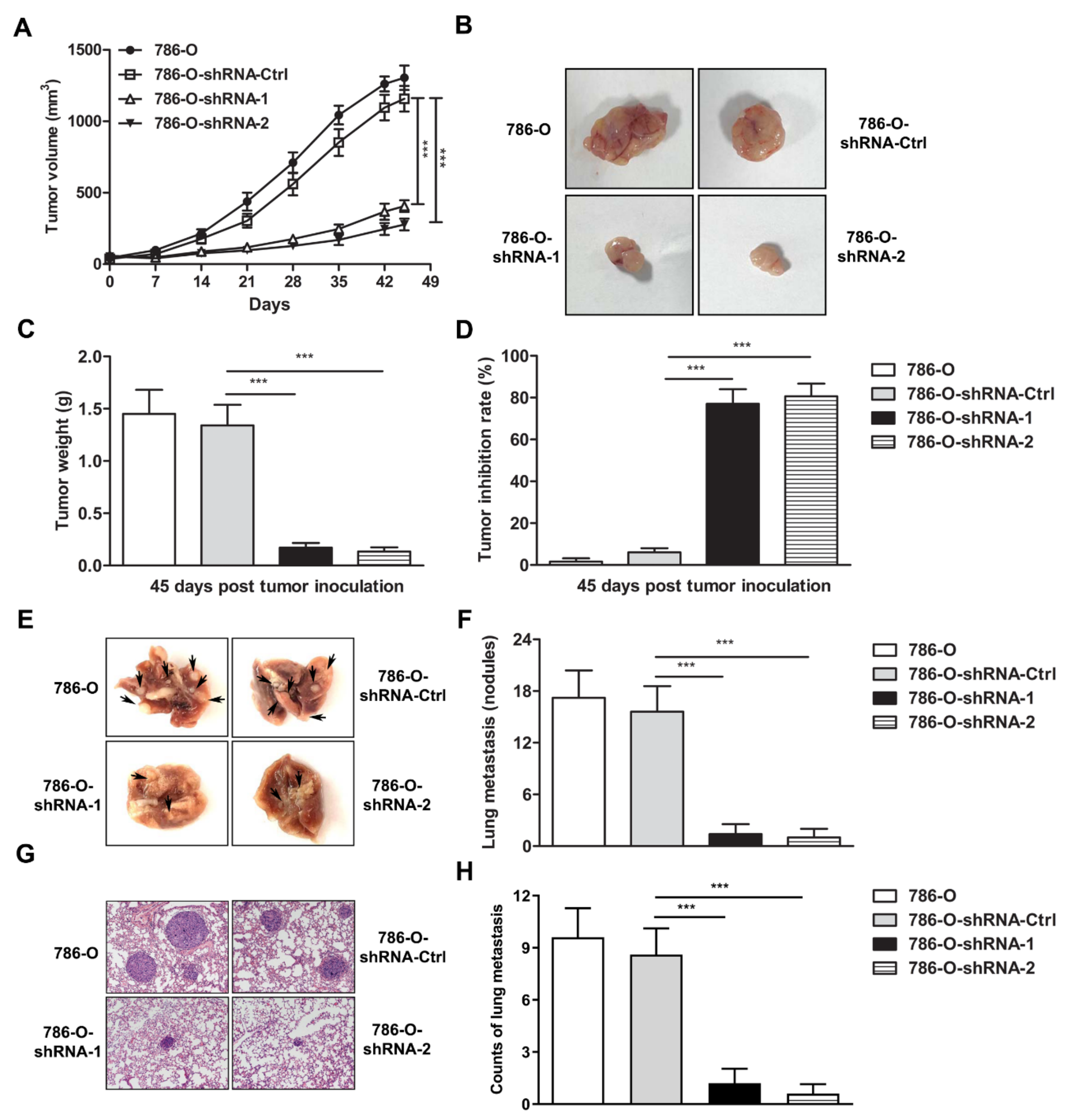
3.3. IFI35 Knockdown Suppresses the Tumor Growth or Metastasis in Subcutaneous or Lung Metastasis Models of Renal Cancer
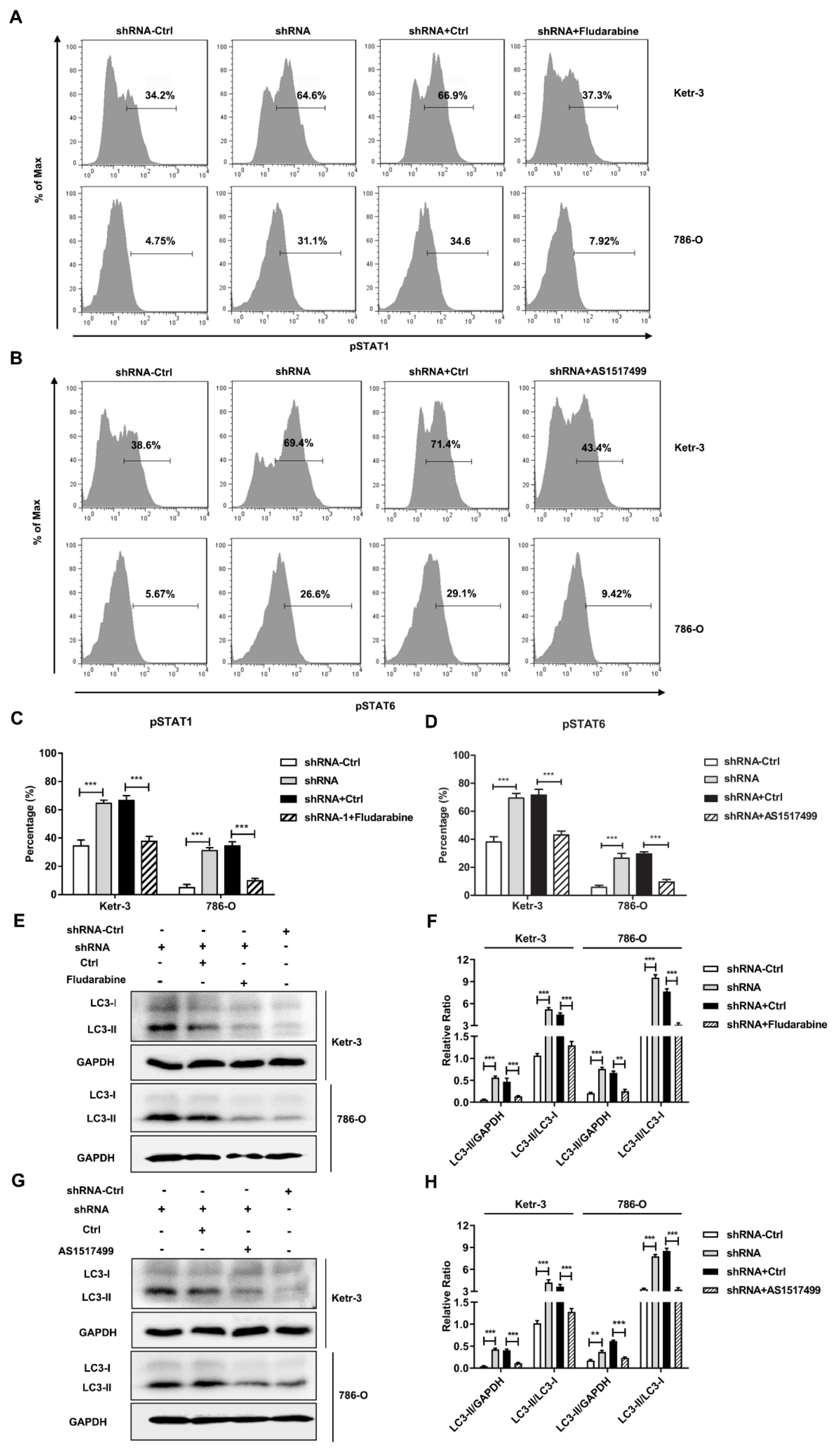
3.4. Knockdown of IFI35 Enhances pSTAT1/pSTAT6 Dependent Autophagy in Renal Cancer Cells
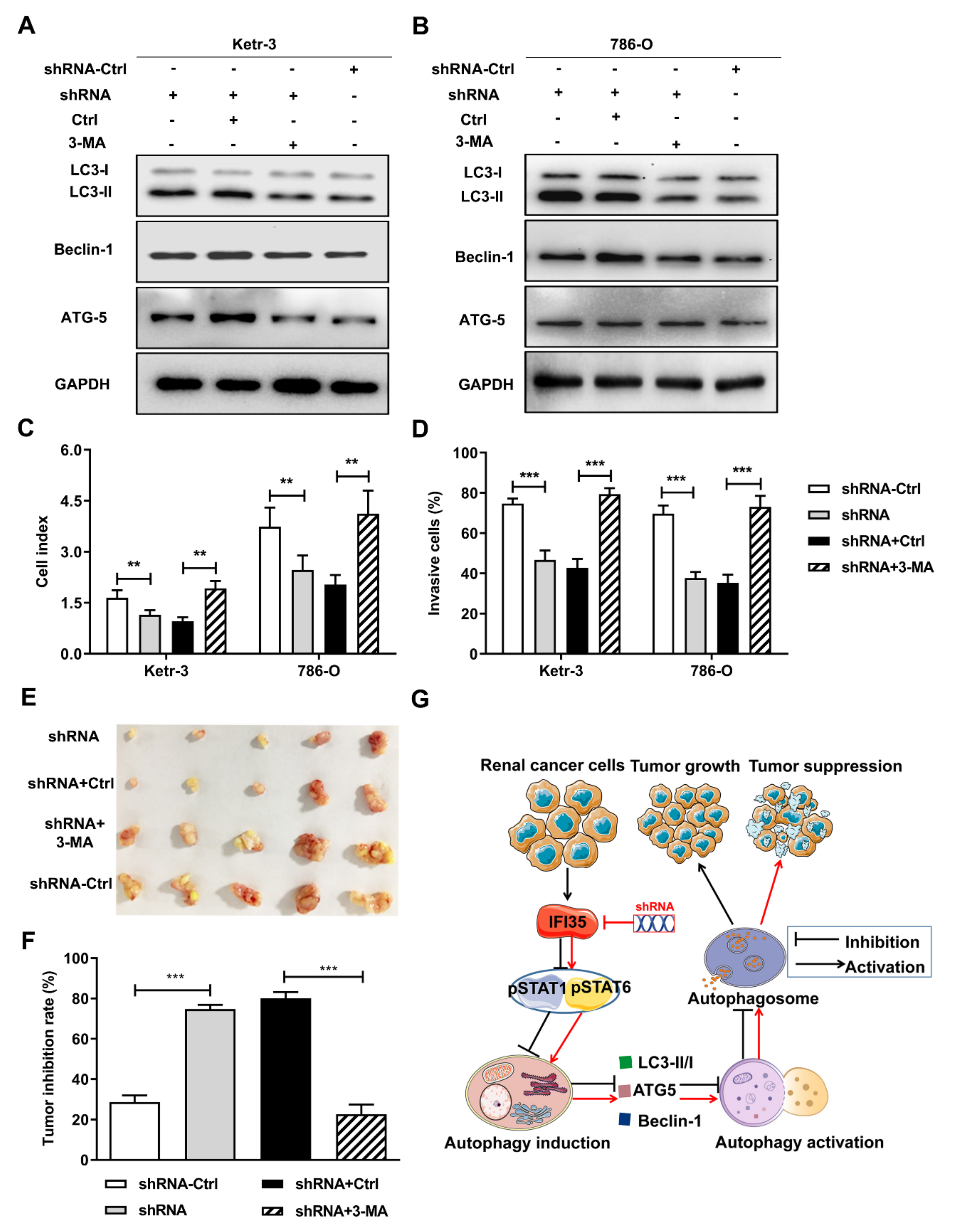
3.5. Autophagy Induced by the Knockdown of IFI35 Is Critical for Inhibiting the Malignant Behavior of Renal Cancer Cells and Tumor Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, H.T.; McGovern, F.J. Renal-cell carcinoma. N. Engl. J. Med. 2005, 353, 2477–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Beltran, A.; Carrasco, J.C.; Cheng, L.; Scarpelli, M.; Kirkali, Z.; Montironi, R. 2009 update on the classification of renal epithelial tumors in adults: Updated classification of renal tumors. Int. J. Urol. 2009, 16, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Marostica, E.; Barber, R.; Denize, T.; Kohane, S.I.; Signoretti, S.; Golden, J.A.; Yu, K.H. Development of a histopathology informatics pipeline for classification and prediction of clinical outcomes in subtypes of renal cell carcinoma. Clin. Cancer Res. 2021, 27, 2868–2878. [Google Scholar] [CrossRef]
- Rini, B.I.; Campbell, S.C.; Escudier, B. Renal cell carcinoma. Lancet 2009, 373, 1119–1132. [Google Scholar] [CrossRef]
- Wahlgren, T.; Harmenberg, U.; Sandstrom, P.; Lundstam, S.; Kowalski, J.; Jakobsson, M.; Sandin, R.; Ljungberg, B. Treatment and overall survival in renal cell carcinoma: A Swedish population-based study (2000-2008). Br. J. Cancer 2013, 108, 1541–1549. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers. 2017, 3, 17009. [Google Scholar] [CrossRef]
- Posadas, E.M.; Limvorasak, S.; Figlin, R.A. Targeted therapies for renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 496–511. [Google Scholar] [CrossRef]
- Atkins, M.B.; Tannir, N.M. Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treat. Rev. 2018, 70, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Barata, P.C.; Rini, B.I. Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J. Clin. 2017, 67, 507–524. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.M.; Carew, J.S.; Nawrocki, S.T. Therapeutic Targeting of Autophagy for Renal Cell Carcinoma Therapy. Cancers 2020, 12, 1185. [Google Scholar] [CrossRef]
- Cao, Q.; Bai, P. Role of Autophagy in Renal Cancer. J. Cancer 2019, 10, 2501–2509. [Google Scholar] [CrossRef] [Green Version]
- Santoni, M.; Piva, F.; De Giorgi, U.; Mosca, A.; Basso, U.; Santini, D.; Buti, S.; Lolli, C.; Terrone, C.; Maruzzo, M.; et al. Autophagic Gene Polymorphisms in Liquid Biopsies and Outcome of Patients with Metastatic Clear Cell Renal Cell Carcinoma. Anticancer Res. 2018, 38, 5773–5782. [Google Scholar] [CrossRef]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef]
- Chai, D.; Shan, H.; Wang, G.; Li, H.; Fang, L.; Song, J.; Zhang, Q.; Bai, J.; Zheng, J. AIM2 is a potential therapeutic target in human renal carcinoma and suppresses its invasion and metastasis via enhancing autophagy induction. Exp. Cell Res. 2018, 370, 561–570. [Google Scholar] [CrossRef]
- Das, A.; Dinh, P.X.; Panda, D.; Pattnaik, A.K. Interferon-inducible protein IFI35 negatively regulates RIG-I antiviral signaling and supports vesicular stomatitis virus replication. J. Viro. 2014, 88, 3103–3113. [Google Scholar] [CrossRef] [Green Version]
- Gounder, A.P.; Yokoyama, C.C.; Jarjour, N.N.; Bricker, T.L.; Edelson, B.T.; Boon, A.C.M. Interferon induced protein 35 exacerbates H5N1 influenza disease through the expression of IL-12p40 homodimer. PLoS Pathog. 2018, 14, e1007001. [Google Scholar] [CrossRef]
- Weix, J.; Haupl, T.; Raio, L.; Villiger, P.M.; Forger, F. The physiologic increase in expression of some type I IFN-inducible genes during pregnancy is not associated with improved disease activity in pregnant patients with rheumatoid arthritis. Transl. Res. 2013, 161, 505–512. [Google Scholar] [CrossRef]
- Xiahou, Z.; Wang, X.; Shen, J.; Zhu, X.; Xu, F.; Hu, R.; Guo, D.; Li, H.; Tian, Y.; Liu, Y.; et al. NMI and IFP35 serve as proinflammatory DAMPs during cellular infection and injury. Nat. Commun. 2017, 8, 950. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, H.; Li, Y.; Dai, X.; Zhou, B.; Li, Q.; Zuo, X.; Luo, H. The role of IFI35 in lupus nephritis and related mechanisms. Mod. Rheumatol. 2017, 27, 1010–1018. [Google Scholar] [CrossRef]
- Imaizumi, T.; Yano, C.; Numata, A.; Tsugawa, K.; Hayakari, R.; Matsumiya, T.; Yoshida, H.; Watanabe, S.; Tsuruga, K.; Kawaguchi, S.; et al. Interferon (IFN)-Induced Protein 35 (IFI35), a Type I Interferon-Dependent Transcript, Upregulates Inflammatory Signaling Pathways by Activating Toll-Like Receptor 3 in Human Mesangial Cells. Kidney Blood Press. Res. 2016, 41, 635–642. [Google Scholar] [CrossRef]
- Bange, F.C.; Vogel, U.; Flohr, T.; Kiekenbeck, M.; Denecke, B.; Bottger, E.C. IFP 35 is an interferon-induced leucine zipper protein that undergoes interferon-regulated cellular redistribution. J. Biol. Chem. 1994, 269, 1091–1098. [Google Scholar] [CrossRef]
- Jian, D.; Wang, W.; Zhou, X.; Jia, Z.; Wang, J.; Yang, M.; Zhao, W.; Jiang, Z.; Hu, X.; Zhu, J. Interferon-induced protein 35 inhibits endothelial cell proliferation, migration and re-endothelialization of injured arteries by inhibiting the nuclear factor-kappa B pathway. Acta. Physiol. 2018, 223, e13037. [Google Scholar] [CrossRef]
- Bai, J.; Mei, P.; Zhang, C.; Chen, F.; Li, C.; Pan, Z.; Liu, H.; Zheng, J. BRG1 is a prognostic marker and potential therapeutic target in human breast cancer. PLoS ONE 2013, 8, e59772. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Jung, J.U. Autophagy genes as tumor suppressors. Curr. Opin. Cell Biol. 2010, 22, 226–233. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wang, X.; Liu, Z.; Wang, Y.; Yin, B.; Yu, P.; Duan, X.; Liao, Z.; Chen, Y.; Liu, C.; et al. SGK1 inhibition induces autophagy-dependent apoptosis via the mTOR-Foxo3a pathway. Br. J. Cancer 2017, 117, 1139–1153. [Google Scholar] [CrossRef]
- Hu, Z.W.; Zhou, L.Q.; Yang, S.; Chen, M.; Yu, H.H.; Tao, R.; Wu, L.J.; Wang, W.; Zhang, Q.; Qin, C.; et al. FTY720 Modulates Microglia Toward Anti-inflammatory Phenotype by Suppressing Autophagy via STAT1 Pathway. Cell Mol. Neurobiol. 2021, 41, 353–364. [Google Scholar] [CrossRef]
- Qin, C.; Liu, Q.; Hu, Z.W.; Zhou, L.Q.; Shang, K.; Bosco, D.B.; Wu, L.J.; Tian, D.S.; Wang, W. Microglial TLR4-dependent autophagy induces ischemic white matter damage via STAT1/6 pathway. Theranostics 2018, 8, 5434–5451. [Google Scholar] [CrossRef]
- Mikami, S.; Oya, M.; Mizuno, R.; Kosaka, T.; Katsube, K.; Okada, Y. Invasion and metastasis of renal cell carcinoma. Med. Mol. Morphol. 2014, 47, 63–67. [Google Scholar] [CrossRef]
- Chen, X.; Li, Z.; Yong, H.; Wang, W.; Wang, D.; Chu, S.; Li, M.; Hou, P.; Zheng, J.; Bai, J. Trim21-mediated HIF-1alpha degradation attenuates aerobic glycolysis to inhibit renal cancer tumorigenesis and metastasis. Cancer Lett. 2021, 508, 115–126. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, X.; Li, H.; Zhang, Y.; Li, X.; Chen, L.; Guo, G.; Gao, Y.; Gu, L.; Xie, Y.; et al. FOXK2 suppresses the malignant phenotype and induces apoptosis through inhibition of EGFR in clear-cell renal cell carcinoma. Int. J. Cancer 2018, 142, 2543–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.L.; Wu, L.W.; Zeng, L.H.; Zhang, Z.Y.; Wang, W.; Zhang, C.; Lin, N.M. ApoC1 promotes the metastasis of clear cell renal cell carcinoma via activation of STAT3. Oncogene 2020, 39, 6203–6217. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.H.; Tian, G. Autophagy as a Vital Therapy Target for Renal Cell Carcinoma. Front. Pharmacol. 2021, 11, 518225. [Google Scholar] [CrossRef]
- Perot, B.P.; Boussier, J.; Yatim, N.; Rossman, J.S.; Ingersoll, M.A.; Albert, M.L. Autophagy diminishes the early interferon-beta response to influenza A virus resulting in differential expression of interferon-stimulated genes. Cell Death Dis. 2018, 9, 539. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Zheng, H.; Dong, W.; Lv, H.; Lin, J.; Guo, K.; Zhang, Y. Antiviral activity of ISG15 against classical swine fever virus replication in porcine alveolar macrophages via inhibition of autophagy by ISGylating BECN1. Vet. Res. 2020, 51, 22. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Zeng, Y.; Tian, X.; Wang, Q.; He, W.; Fan, J.; Gou, X. Attenuation of everolimus-induced cytotoxicity by a protective autophagic pathway involving ERK activation in renal cell carcinoma cells. Drug Des. Devel. Ther. 2018, 12, 911–920. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Niso-Santano, M.; Adjemian, S.; Takehara, T.; Malik, S.A.; Minoux, H.; Souquere, S.; Mariño, G.; Lachkar, S.; Senovilla, L.; et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol. Cell. 2012, 48, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Liu, Y.; Chen, J.; Lin, H.; Guo, H.; Wu, Y.; Xu, Y.; Zhou, Y.; Zhou, W.; Lu, R.; et al. JAK/STAT pathway promotes the progression of diabetic kidney disease via autophagy in podocytes. Eur. J. Pharmacol. 2021, 902, 174121. [Google Scholar] [CrossRef]
- Shirai, K.; Shimada, T.; Yoshida, H.; Hayakari, R.; Matsumiya, T.; Tanji, K.; Murakami, M.; Tanaka, H.; Imaizumi, T. Interferon (IFN)-induced protein 35 (IFI35) negatively regulates IFN-beta-phosphorylated STAT1-RIG-I-CXCL10/CCL5 axis in U373MG astrocytoma cells treated with polyinosinic-polycytidylic acid. Brain Res. 2017, 1658, 60–67. [Google Scholar] [CrossRef]
- Papageorgis, P.; Ozturk, S.; Lambert, A.W.; Neophytou, C.M.; Tzatsos, A.; Wong, C.K.; Thiagalingam, S.; Constantinou, A.I. Targeting IL13Ralpha2 activates STAT6-TP63 pathway to suppress breast cancer lung metastasis. Breast Cancer Res. 2015, 17, 98. [Google Scholar] [CrossRef] [Green Version]
- Leon-Cabrera, S.A.; Molina-Guzman, E.; Delgado-Ramirez, Y.G.; Vazquez-Sandoval, A.; Ledesma-Soto, Y.; Perez-Plasencia, C.G.; Chirino, Y.I.; Delgado-Buenrostro, N.L.; Rodriguez-Sosa, M.; Vaca-Paniagua, F.; et al. Lack of STAT6 Attenuates Inflammation and Drives Protection against Early Steps of Colitis-Associated Colon Cancer. Cancer Immunol. Res. 2017, 5, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Ho, H.H.; Ivashkiv, L.B. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem. 2006, 281, 14111–14118. [Google Scholar] [CrossRef] [Green Version]
- Schindler, C.; Darnell, J.J. Transcriptional responses to polypeptide ligands: The JAK-STAT pathway. Annu. Rev. Biochem. 1995, 64, 621–651. [Google Scholar] [CrossRef]
- Botbol, Y.; Patel, B.; Macian, F. Common γ-chain cytokine signaling is required for macroautophagy induction during CD4+ T-cell activation. Autophagy 2015, 11, 1864–1877. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | IFI35 Staining | |||
|---|---|---|---|---|
| Low (%) | High (%) | Total | p * | |
| Age | ||||
| >56 years | 40 (42.4) | 68 (57.6) | 113 | 0.761 |
| ≤56 years | 36 (47.3) | 56 (52.7) | 87 | |
| Gender | ||||
| Male | 52 (56.7) | 80 (43.1) | 132 | 0.571 |
| Female | 24 (54.3) | 44 (45.7) | 68 | |
| Tumor size | ||||
| ≤7 cm | 56 (54.6) | 98 (45.4) | 154 | 0.383 |
| >7 cm | 20 (53.8) | 26 (46.2) | 46 | |
| Histology Grade | ||||
| I–III | 67 (72.2) | 114 (27.8) | 181 | 0.376 |
| IV | 9 (54.2) | 10 (45.8) | 19 | |
| pT status | ||||
| pT1–pT3 | 67 (62.1) | 103 (37.9) | 187 | 0.677 |
| pT4 | 4 (33.3) | 8 (66.7) | 13 | |
| pN status | ||||
| N0 | 63 (57.0) | 109 (43.0) | 172 | 0.639 |
| N1–N3 | 8 (54.2) | 11 (45.8) | 19 | |
| pM status | ||||
| M0 | 68 (51.8) | 98 (49.2) | 166 | 0.135 |
| M | 2 (66.7) | 9 (33.3) | 9 | |
| TNM stage | ||||
| I–III | 54 (59.6) | 76 (41.4) | 160 | <0.05 |
| IV | 1 (75.0) | 10 (25.0) | 9 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, D.; Shi, S.Y.; Sobhani, N.; Ding, J.; Zhang, Z.; Jiang, N.; Wang, G.; Li, M.; Li, H.; Zheng, J.; et al. IFI35 Promotes Renal Cancer Progression by Inhibiting pSTAT1/pSTAT6-Dependent Autophagy. Cancers 2022, 14, 2861. https://doi.org/10.3390/cancers14122861
Chai D, Shi SY, Sobhani N, Ding J, Zhang Z, Jiang N, Wang G, Li M, Li H, Zheng J, et al. IFI35 Promotes Renal Cancer Progression by Inhibiting pSTAT1/pSTAT6-Dependent Autophagy. Cancers. 2022; 14(12):2861. https://doi.org/10.3390/cancers14122861
Chicago/Turabian StyleChai, Dafei, Shang Yuchen Shi, Navid Sobhani, Jiage Ding, Zichun Zhang, Nan Jiang, Gang Wang, Minle Li, Hailong Li, Junnian Zheng, and et al. 2022. "IFI35 Promotes Renal Cancer Progression by Inhibiting pSTAT1/pSTAT6-Dependent Autophagy" Cancers 14, no. 12: 2861. https://doi.org/10.3390/cancers14122861