
MALAT1 as a Regulator of the Androgen-Dependent Choline Kinase A Gene in the Metabolic Rewiring of Prostate Cancer

 ,
,  ,
,  ,
,  ,
, 
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
- hCHKA 5′-GGTCACTTGGGCCAAAACTC-3’ and 5′-GCCGGCTCGGGATGA-3′;
- hPLCG1 5′-GCTTCTATGTAGAGGCAAACCCTATG-3′ and 5′-CCCTCTGGGCCTTGTAGTCA-3′
- hSMPD1 5′-TGCCCAATCTGCAAAGGTCTA-3′ and 5′-GCCACATTGGGTTCCTTCTTC-3′
- hSGMS1 5′-AGCATGATTCAGGCACACCAT-3′ and 5′-TCATGTTTCCCAACCAGACACT-3′
- hCERK 5′-TGGCACCACTGTTCACCTTA-3′ and 5′-CTCCTTGGCCTGATTAGCAT-3′
- CHKAprom (−3500) 5′-GGAAAAGGTTTGGTAATTGGAACA-3′ and 5′-CTGTGCACAAGTAGACGAGTTTGA-3′
- CHKAprom (−1500) 5′-CGCCACAGCAGCCTTACAA-3′ and 5′-CCCAAAGTGCTGGGATTACAG-3′
- CHKAprom (Intron 2, 37,500) 5′-CAGGACTTAGGGAGCCTGAACA-3′ and 5′-GGCAAGATGGACTTCTGCAATAT-3′.
- hCHKA (Intron 2, 27,500) 5′-GATGGGAGTAATGGAGGGTTCTG-3′ and 5′-CGTTAGTGACATGTGGCTGATGA-3′
- CERKprom (TSS) 5′-ACAAGACGGACTGTGGATGGA-3′ and 5′-CATCTGTTCTTGGAGTAAACTGCAA-3′
- CERKprom (−8600) 5′-GCCACTCTGTTCTGCGATCAC-3′ and 5′-CTGTTGGAGCCTCCGTTTTC-3′
- CERKprom (Intron, 46,500) 5′-CTTGGGAGACGGGTCTTCTG-3′ and 5′-GGTTGGTGTGCCTGATGAGA-3′
3. Results
3.1. MALAT1 Targeting Changes Metabolites of the Choline Pathway in PCa Cells
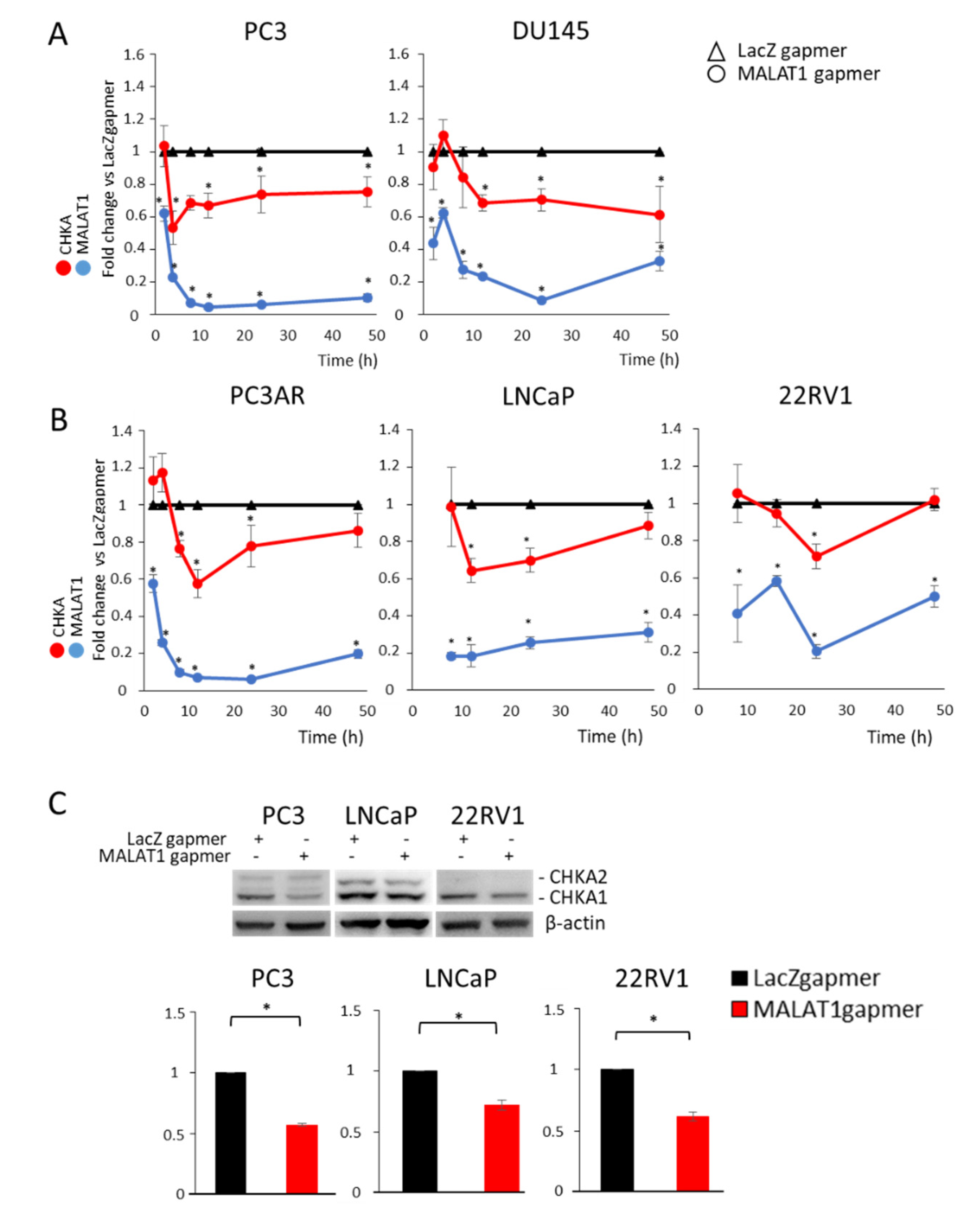
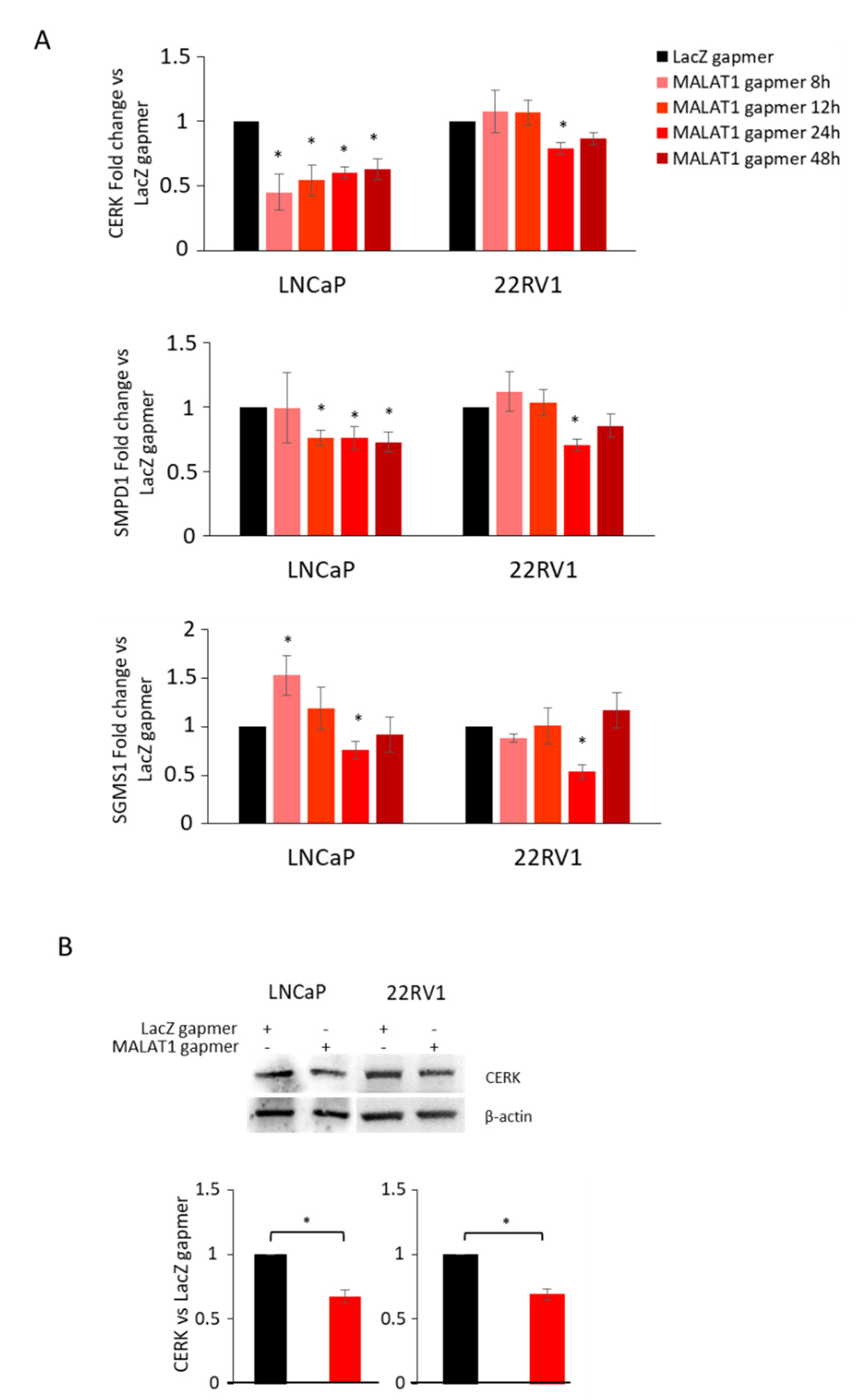
3.2. MALAT1 Targeting Decreases CHKA Gene Expression in PCa Cells
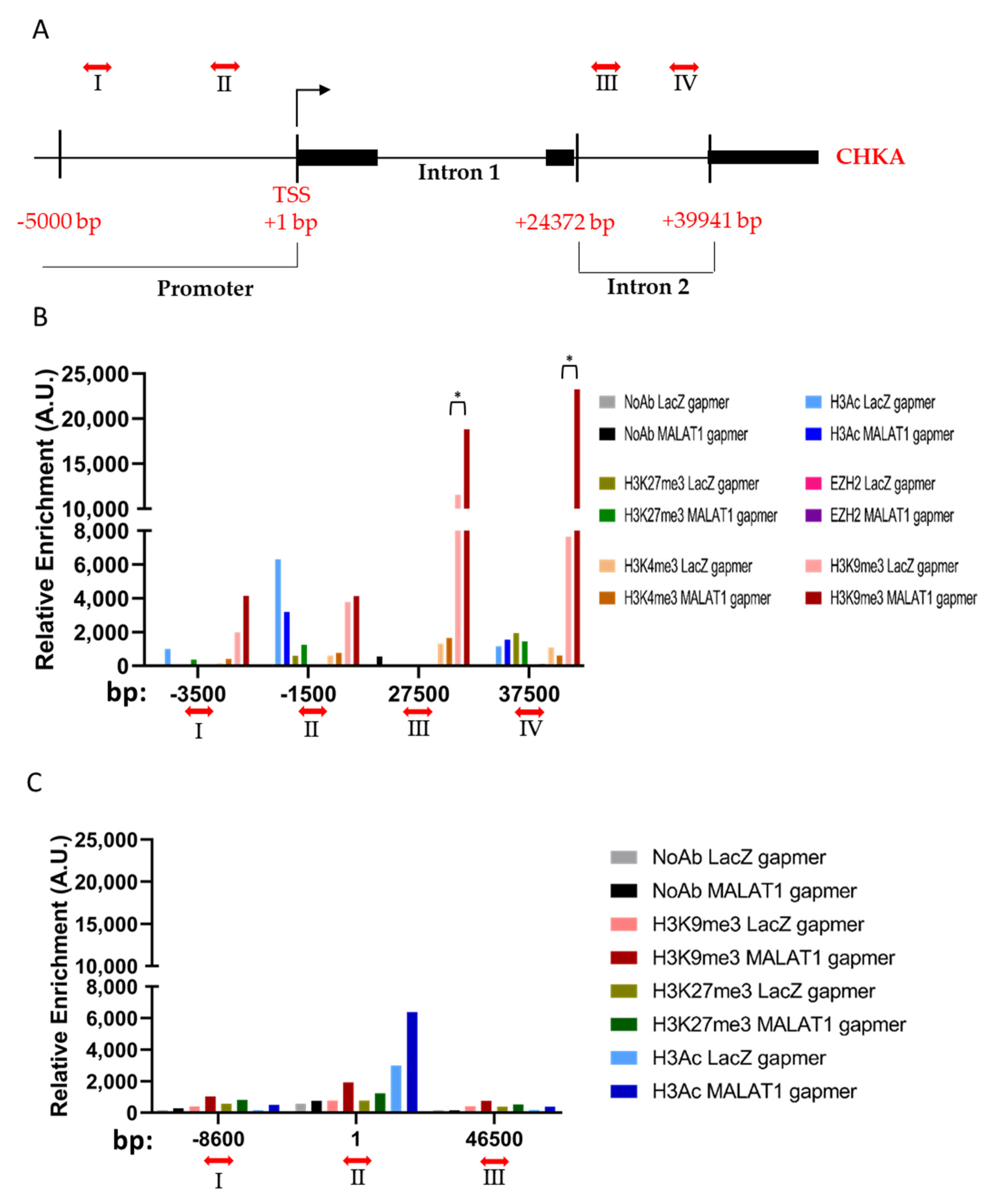
3.3. MALAT1 Targeting Determines Chromatin Remodeling on CHKA Genomic Region
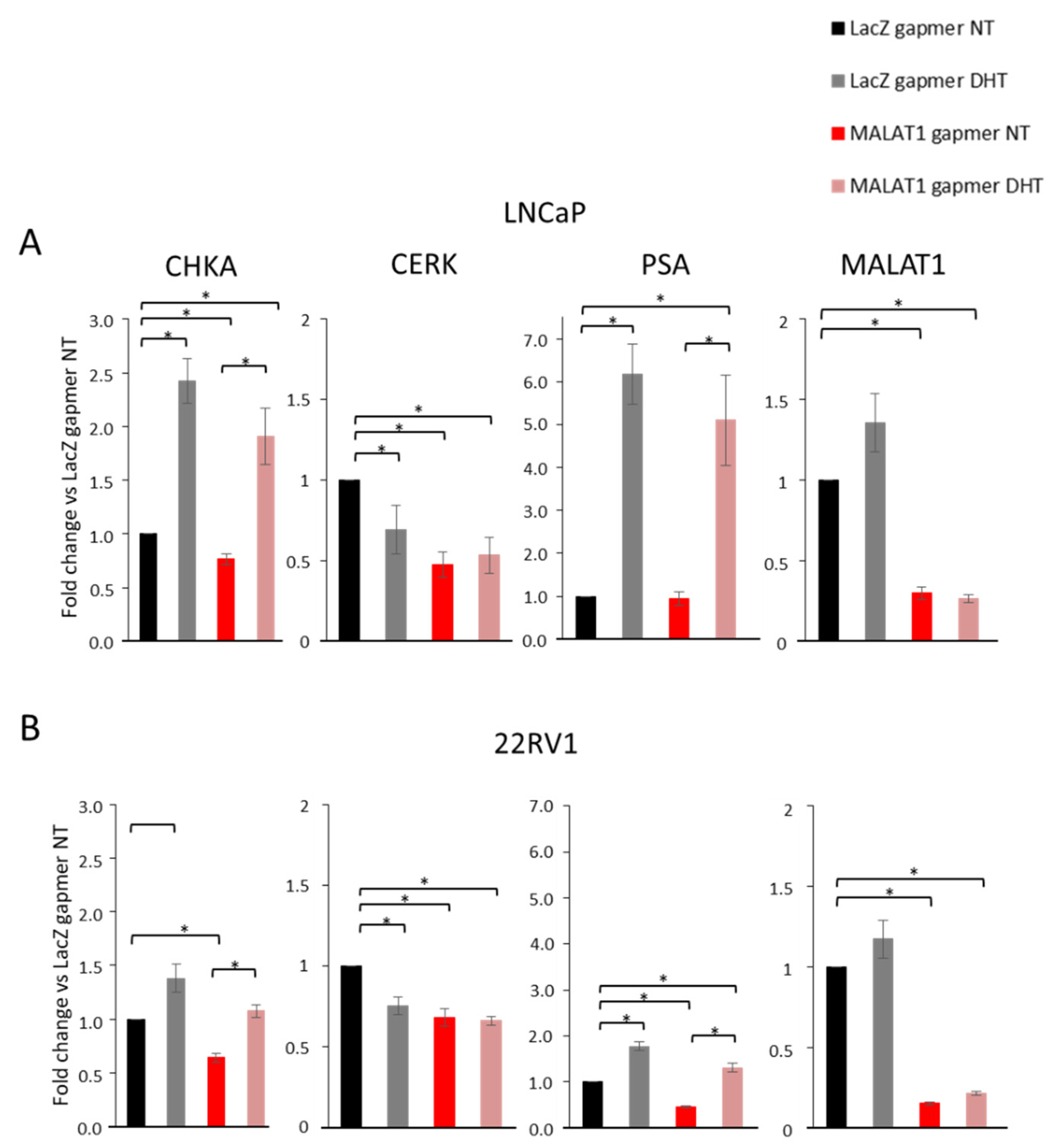
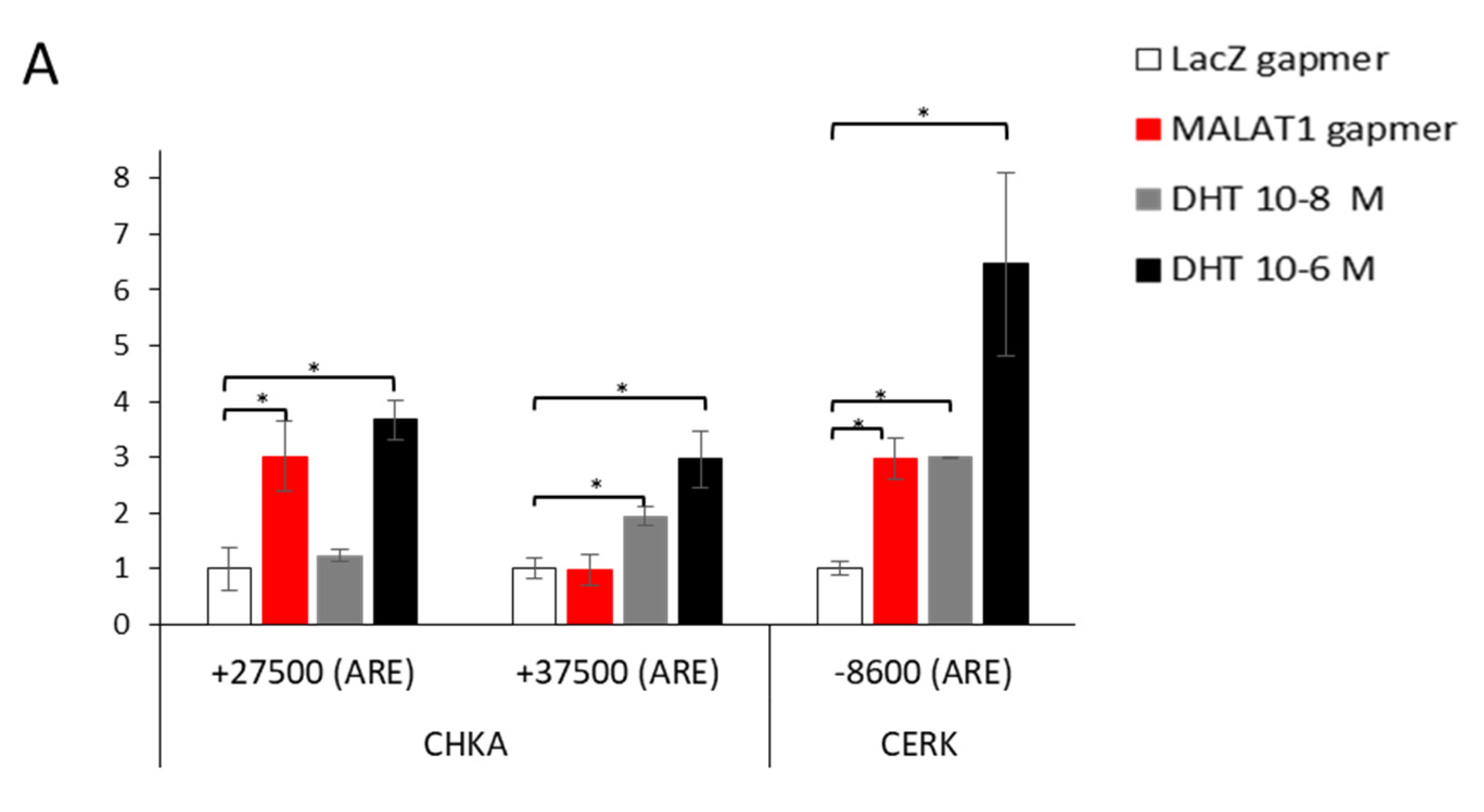
3.4. Effects of MALAT1 Targeting Combined with Dihydrotestosterone (DHT) Treatment on Metabolic Genes Involved in Phospholipid/Sphingolipid Homeostasis
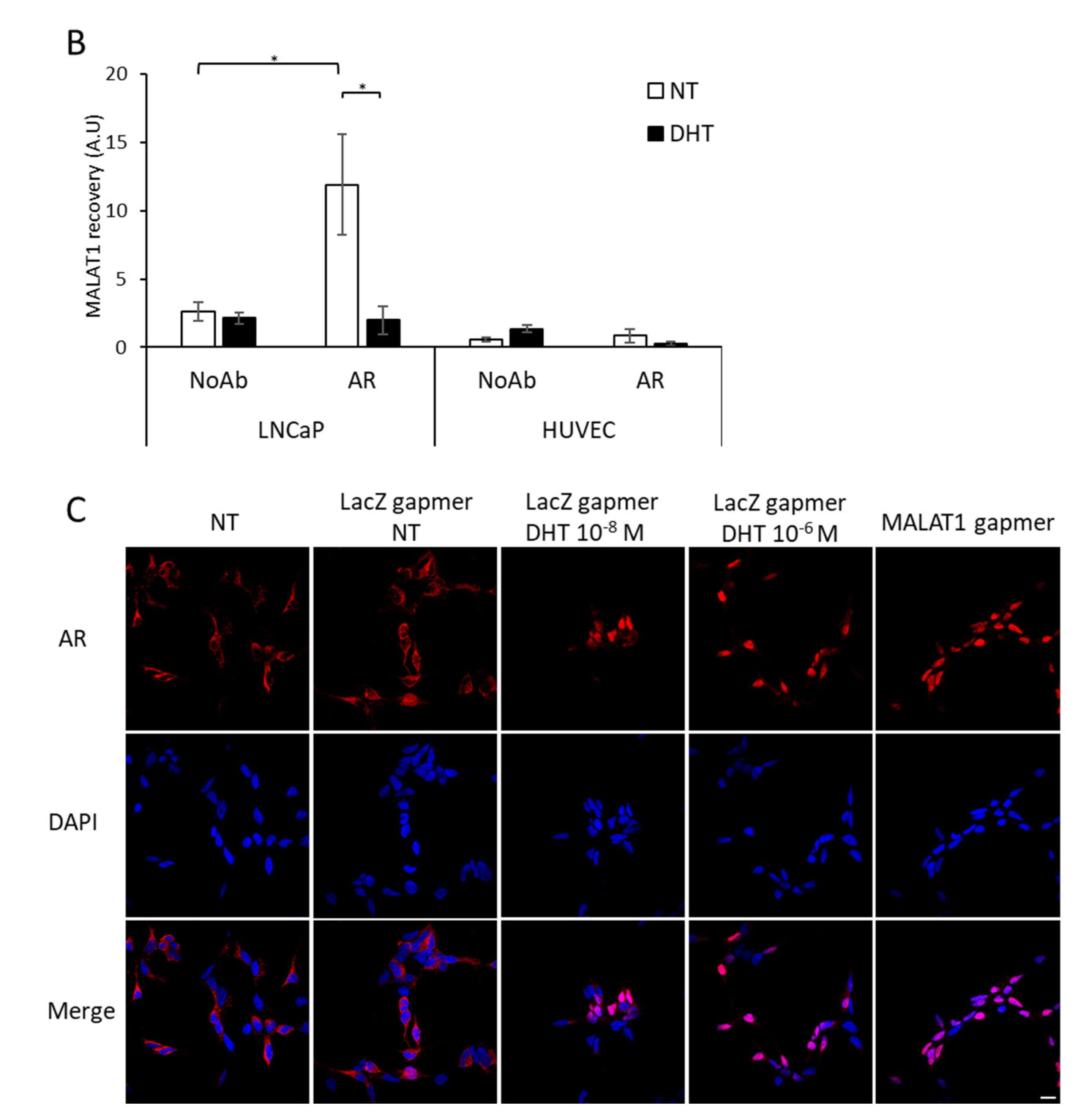
3.5. Dynamic Recruitment of AR on CHKA Genomic Sequences before/after MALAT1 Targeting in the Presence or Absence of DHT
4. Discussion
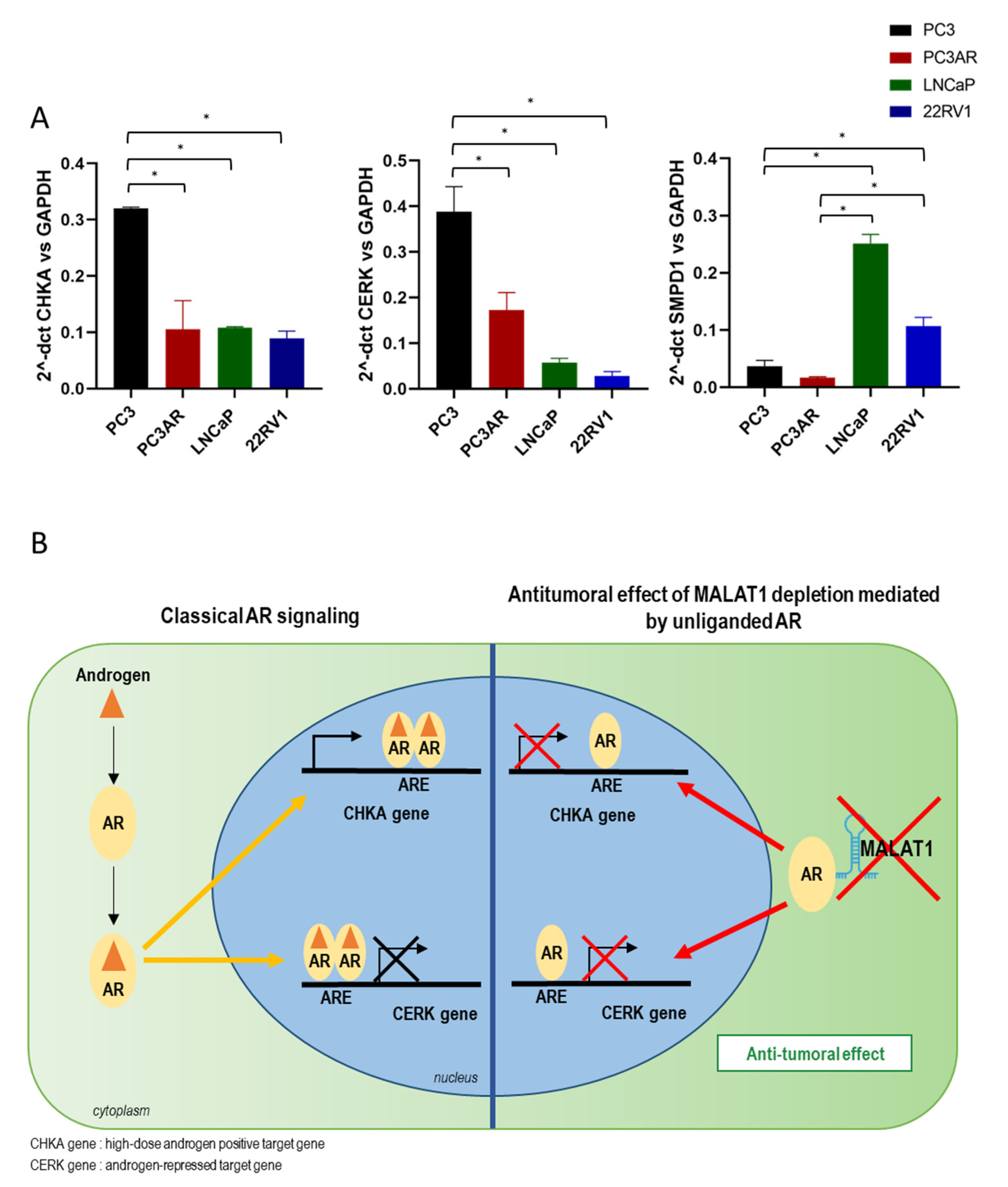
4.1. CHKA Is a Target of MALAT1
4.2. Lipid Metabolism Is under the Control of AR Signaling
4.3. Crosstalk between MALAT1/AR and AR Recruitment along CHKA Genomic Regions before/after MALAT1 Depletion +/− DHT
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hashim, D.; Carioli, G.; Malvezzi, M.; Bertuccio, P.; Waxman, S.; Negri, E.; La Vecchia, C.; Boffetta, P. Cancer Mortality in the Oldest Old: A Global Overview. Aging 2020, 12, 16744–16758. [Google Scholar] [CrossRef]
- Peltomaa, A.I.; Raittinen, P.; Talala, K.; Taari, K.; Tammela, T.L.J.; Auvinen, A.; Murtola, T.J. Prostate Cancer Prognosis after Initiation of Androgen Deprivation Therapy among Statin Users. A Population-Based Cohort Study. Prostate Cancer Prostatic Dis. 2021, 24, 917–924. [Google Scholar] [CrossRef]
- Kopecký, J.; Navláčilová, V.; Janoutová, J.; Janout, V. Epidemiological Study on More Accurate Diagnosis of Prostate Cancer. Cent. Eur. J. Public Health 2020, 28, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Massie, C.E.; Neal, D.E. Kinase Joins the Chaperone Club: Androgen-Regulated Kinome Reveals Choline Kinase Alpha as a Potential Drug Target in Prostate Cancer. Mol. Cell. Oncol. 2016, 3, e1140262. [Google Scholar] [CrossRef] [Green Version]
- Asim, M.; Massie, C.E.; Orafidiya, F.; Pértega-Gomes, N.; Warren, A.Y.; Esmaeili, M.; Selth, L.A.; Zecchini, H.I.; Luko, K.; Qureshi, A.; et al. Choline Kinase Alpha as an Androgen Receptor Chaperone and Prostate Cancer Therapeutic Target. J. Natl. Cancer Inst. 2016, 108, djv371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, C.; Liao, H.; Ishidate, K. Structure and Function of Choline Kinase Isoforms in Mammalian Cells. Prog. Lipid Res. 2004, 43, 266–281. [Google Scholar] [CrossRef]
- Chen, X.; Qiu, H.; Wang, C.; Yuan, Y.; Tickner, J.; Xu, J.; Zou, J. Molecular Structure and Differential Function of Choline Kinases CHKα and CHKβ in Musculoskeletal System and Cancer. Cytokine Growth Factor Rev. 2017, 33, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Bian, J.; Wang, L.; Zhou, J.-Y.; Wang, Y.; Zhao, Y.; Wu, L.-L.; Hu, J.-J.; Li, B.; Chen, S.-J.; et al. Dysregulated Choline Metabolism in T-Cell Lymphoma: Role of Choline Kinase-α and Therapeutic Targeting. Blood Cancer J. 2015, 5, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernando, E.; Sarmentero-Estrada, J.; Koppie, T.; Belda-Iniesta, C.; Ramírez de Molina, V.; Cejas, P.; Ozu, C.; Le, C.; Sánchez, J.J.; González-Barón, M.; et al. A Critical Role for Choline Kinase-Alpha in the Aggressiveness of Bladder Carcinomas. Oncogene 2009, 28, 2425–2435. [Google Scholar] [CrossRef] [Green Version]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline Metabolism in Malignant Transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, C. Regulation of Phosphatidylcholine Biosynthesis. Prog. Lipid Res. 1991, 29, 87–105. [Google Scholar] [CrossRef]
- Ramírez de Molina, A.; Báñez-Coronel, M.; Gutiérrez, R.; Rodríguez-González, A.; Olmeda, D.; Megías, D.; Lacal, J.C. Choline Kinase Activation Is a Critical Requirement for the Proliferation of Primary Human Mammary Epithelial Cells and Breast Tumor Progression. Cancer Res. 2004, 64, 6732–6739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janardhan, S.; Srivani, P.; Sastry, G.N. Choline Kinase: An Important Target for Cancer. Curr. Med. Chem. 2006, 13, 1169–1186. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Cherukuri, M.K.; Choyke, P.L. Metabolic Reprogramming in Prostate Cancer. Br. J. Cancer 2021, 125, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Jabalera, Y.; Sola-Leyva, A.; Peigneux, A.; Vurro, F.; Iglesias, G.R.; Vilchez-Garcia, J.; Pérez-Prieto, I.; Aguilar-Troyano, F.J.; López-Cara, L.C.; Carrasco-Jiménez, M.P.; et al. Biomimetic Magnetic Nanocarriers Drive Choline Kinase Alpha Inhibitor inside Cancer Cells for Combined Chemo-Hyperthermia Therapy. Pharmaceutics 2019, 11, 408. [Google Scholar] [CrossRef] [Green Version]
- Mariotto, E.; Viola, G.; Ronca, R.; Persano, L.; Aveic, S.; Bhujwalla, Z.M.; Mori, N.; Accordi, B.; Serafin, V.; López-Cara, L.C.; et al. Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer. Cancers 2018, 10, 391. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Brickute, D.; Braga, M.; Barnes, C.; Lu, H.; Allott, L.; Aboagye, E.O. Novel Non-Congeneric Derivatives of the Choline Kinase Alpha Inhibitor ICL-CCIC-0019. Pharmaceutics 2021, 13, 1078. [Google Scholar] [CrossRef] [PubMed]
- Nanni, S.; Aiello, A.; Salis, C.; Re, A.; Cencioni, C.; Bacci, L.; Pierconti, F.; Pinto, F.; Ripoli, C.; Ostano, P.; et al. Metabolic Reprogramming by Malat1 Depletion in Prostate Cancer. Cancers 2021, 13, 15. [Google Scholar] [CrossRef]
- Aiello, A.; Bacci, L.; Re, A.; Ripoli, C.; Pierconti, F.; Pinto, F.; Masetti, R.; Grassi, C.; Gaetano, C.; Bassi, P.F.; et al. MALAT1 and HOTAIR Long Non-Coding RNAs Play Opposite Role in Estrogen-Mediated Transcriptional Regulation in Prostate Cancer Cells. Sci. Rep. 2016, 6, 38414. [Google Scholar] [CrossRef]
- Bacci, L.; Aiello, A.; Ripoli, C.; Loria, R.; Pugliese, D.; Pierconti, F.; Rotili, D.; Strigari, L.; Pinto, F.; Bassi, P.F.; et al. H19-Dependent Transcriptional Regulation of Β3 and Β4 Integrins Upon Estrogen and Hypoxia Favors Metastatic Potential in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 4012. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Huang, C.-H. LASAGNA-Search: An Integrated Web Tool for Transcription Factor Binding Site Search and Visualization. Biotechniques 2013, 54, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The Androgen Receptor Fuels Prostate Cancer by Regulating Central Metabolism and Biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.; Qi, J.; Filipp, F.V. Refinement of the Androgen Response Element Based on ChIP-Seq in Androgen-Insensitive and Androgen-Responsive Prostate Cancer Cell Lines. Sci. Rep. 2016, 6, 32611. [Google Scholar] [CrossRef] [PubMed]
- Camacho, L.; Zabala-Letona, A.; Cortazar, A.R.; Astobiza, I.; Dominguez-Herrera, A.; Ercilla, A.; Crespo, J.; Viera, C.; Fernández-Ruiz, S.; Martinez-Gonzalez, A.; et al. Identification of Androgen Receptor Metabolic Correlome Reveals the Repression of Ceramide Kinase by Androgens. Cancers 2021, 13, 4307. [Google Scholar] [CrossRef] [PubMed]
- Saulle, E.; Spinello, I.; Quaranta, M.T.; Pasquini, L.; Pelosi, E.; Iorio, E.; Castelli, G.; Chirico, M.; Pisanu, M.E.; Ottone, T.; et al. Targeting Lactate Metabolism by Inhibiting MCT1 or MCT4 Impairs Leukemic Cell Proliferation, Induces Two Different Related Death-Pathways and Increases Chemotherapeutic Sensitivity of Acute Myeloid Leukemia Cells. Front. Oncol. 2020, 10, 621458. [Google Scholar] [CrossRef] [PubMed]
- Metere, A.; Graves, C.E.; Chirico, M.; Caramujo, M.J.; Pisanu, M.E.; Iorio, E. Metabolomic Reprogramming Detected by 1H-NMR Spectroscopy in Human Thyroid Cancer Tissues. Biology 2020, 9, 112. [Google Scholar] [CrossRef]
- Iorio, E.; Ricci, A.; Bagnoli, M.; Pisanu, M.E.; Castellano, G.; Di Vito, M.; Venturini, E.; Glunde, K.; Bhujwalla, Z.M.; Mezzanzanica, D.; et al. Activation of Phosphatidylcholine Cycle Enzymes in Human Epithelial Ovarian Cancer Cells. Cancer Res. 2010, 70, 2126–2135. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Lin, C.; Jin, C.; Yang, J.C.; Tanasa, B.; Li, W.; Merkurjev, D.; Ohgi, K.A.; Meng, D.; Zhang, J.; et al. LncRNA-Dependent Mechanisms of Androgen-Receptor-Regulated Gene Activation Programs. Nature 2013, 500, 598–602. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Ma, J.; Pan, X.; Zhao, L.; Wu, Y.; Lin, H.; Zhao, Y.; Jiang, H.; Pan, T.; Li, X.; et al. Crosstalk of FGFR1 Signaling and Choline Metabolism Promotes Cell Proliferation and Survival in Prostate Cancer Cells. Int. J. Cancer 2022, 150, 1525–1536. [Google Scholar] [CrossRef]
- Granata, A.; Nicoletti, R.; Perego, P.; Iorio, E.; Krishnamachary, B.; Benigni, F.; Ricci, A.; Podo, F.; Bhujwalla, Z.M.; Canevari, S.; et al. Global Metabolic Profile Identifies Choline Kinase Alpha as a Key Regulator of Glutathione-Dependent Antioxidant Cell Defense in Ovarian Carcinoma. Oncotarget 2015, 6, 11216–11230. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.; Satta, A.; Garrone, G.; Cavalleri, A.; Napoli, A.; Raspagliesi, F.; Figini, M.; De Cecco, L.; Iorio, E.; Tomassetti, A.; et al. Choline Kinase Alpha Impairment Overcomes TRAIL Resistance in Ovarian Cancer Cells. J. Exp. Clin. Cancer Res. 2021, 40, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y.; Guan, X.; Li, X.; Zhao, Z.; Gao, Y.; Zhang, X.; Chen, R. An Integrated Transcriptomics and Proteomics Analysis Implicates LncRNA MALAT1 in the Regulation of Lipid Metabolism. Mol. Cell. Proteom. 2021, 20, 100141. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and Their Metabolism in Physiology and Disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Podo, F.; Canevari, S.; Canese, R.; Pisanu, M.E.; Ricci, A.; Iorio, E. MR Evaluation of Response to Targeted Treatment in Cancer Cells. NMR Biomed. 2011, 24, 648–672. [Google Scholar] [CrossRef]
- Zou, Y.; Huang, L.; Sun, S.; Yue, F.; Li, Z.; Ma, Y.; Ma, H. Choline Kinase Alpha Promoted Glioma Development by Activating PI3K/AKT Signaling Pathway. Cancer Biother. Radiopharm. 2021, 16, 1–9. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Popov, A.V.; Delikatny, E.J. Choline Kinase Alpha-Putting the ChoK-Hold on Tumor Metabolism. Prog. Lipid Res. 2016, 63, 28–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adada, M.; Luberto, C.; Canals, D. Inhibitors of the Sphingomyelin Cycle: Sphingomyelin Synthases and Sphingomyelinases. Chem. Phys. Lipids 2016, 197, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Okazaki, T. Role of Ceramide/Sphingomyelin (SM) Balance Regulated through “SM Cycle” in Cancer. Cell. Signal. 2021, 87, 110119. [Google Scholar] [CrossRef]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de Novo Lipogenesis Targets Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Bhujwalla, Z.M.; Glunde, K. Targeting Phospholipid Metabolism in Cancer. Front. Oncol. 2016, 6, 266. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Martino, S.; Iorio, E.; Cencioni, C.; Aiello, A.; Spallotta, F.; Chirico, M.; Pisanu, M.E.; Grassi, C.; Pontecorvi, A.; Gaetano, C.; et al. MALAT1 as a Regulator of the Androgen-Dependent Choline Kinase A Gene in the Metabolic Rewiring of Prostate Cancer. Cancers 2022, 14, 2902. https://doi.org/10.3390/cancers14122902
De Martino S, Iorio E, Cencioni C, Aiello A, Spallotta F, Chirico M, Pisanu ME, Grassi C, Pontecorvi A, Gaetano C, et al. MALAT1 as a Regulator of the Androgen-Dependent Choline Kinase A Gene in the Metabolic Rewiring of Prostate Cancer. Cancers. 2022; 14(12):2902. https://doi.org/10.3390/cancers14122902
Chicago/Turabian StyleDe Martino, Sara, Egidio Iorio, Chiara Cencioni, Aurora Aiello, Francesco Spallotta, Mattea Chirico, Maria Elena Pisanu, Claudio Grassi, Alfredo Pontecorvi, Carlo Gaetano, and et al. 2022. "MALAT1 as a Regulator of the Androgen-Dependent Choline Kinase A Gene in the Metabolic Rewiring of Prostate Cancer" Cancers 14, no. 12: 2902. https://doi.org/10.3390/cancers14122902
APA StyleDe Martino, S., Iorio, E., Cencioni, C., Aiello, A., Spallotta, F., Chirico, M., Pisanu, M. E., Grassi, C., Pontecorvi, A., Gaetano, C., Nanni, S., & Farsetti, A. (2022). MALAT1 as a Regulator of the Androgen-Dependent Choline Kinase A Gene in the Metabolic Rewiring of Prostate Cancer. Cancers, 14(12), 2902. https://doi.org/10.3390/cancers14122902