
Proteomic Profiling Identifies Co-Regulated Expression of Splicing Factors as a Characteristic Feature of Intravenous Leiomyomatosis

 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
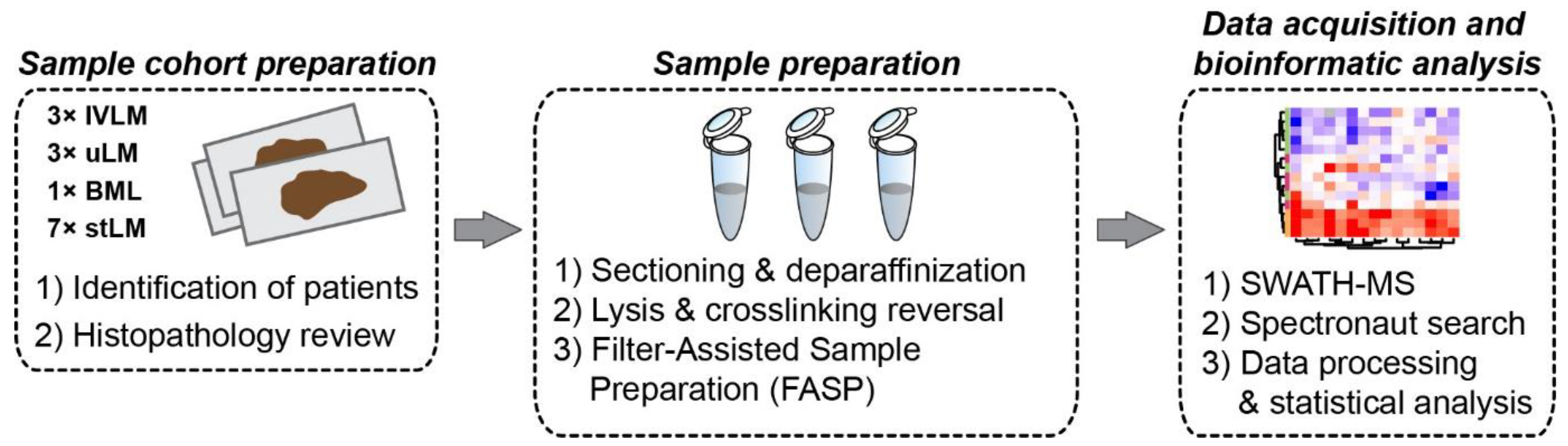
2. Materials and Methods
2.1. Patients and Tumour Specimens
2.2. Protein Extraction and Sample Preparation
2.3. SWATH-MS Data Acquisition and Processing
2.4. Data Processing and Statistical Methods
2.5. Immunohistochemical Staining and Scoring
3. Results
3.1. Quantitative Proteomic Profiling of Smooth Muscle Tumours
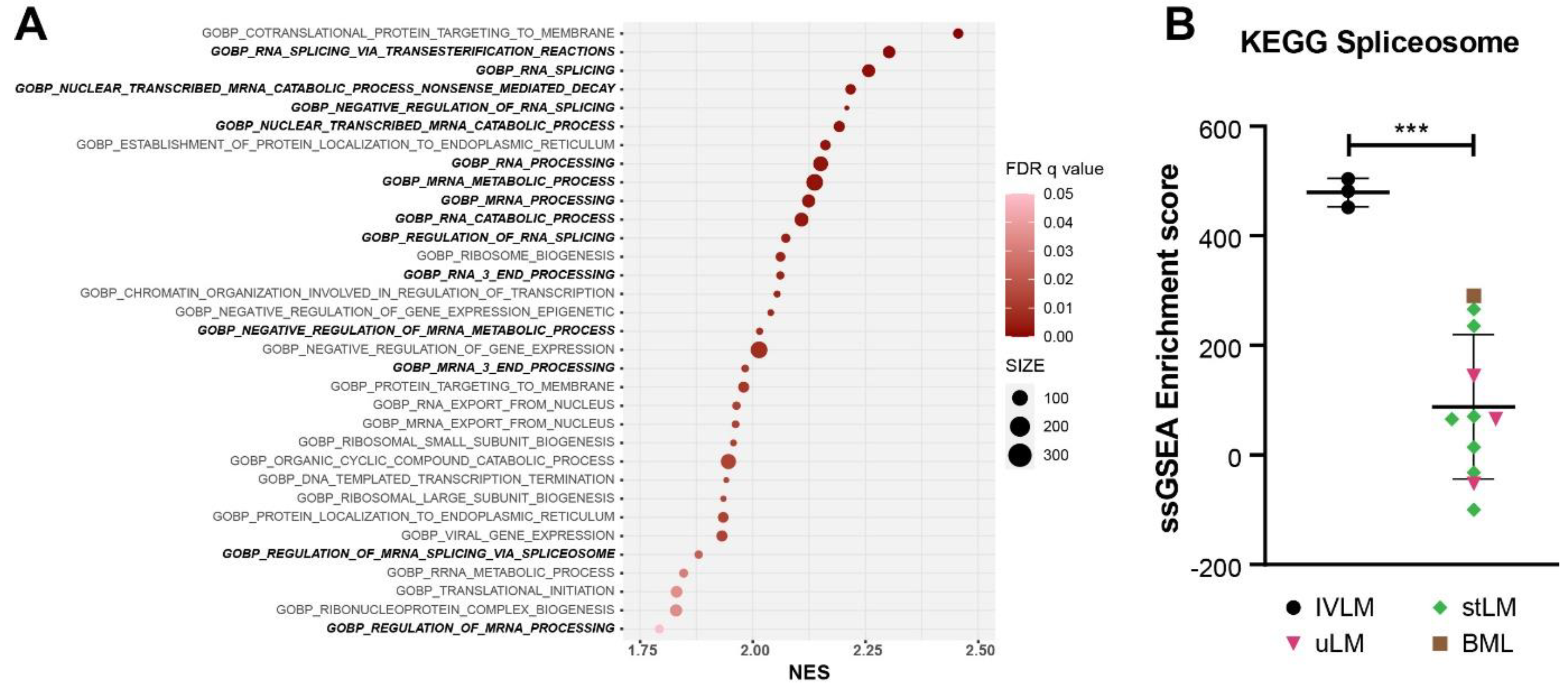
3.2. Enrichment of Splicing Processes in IVLM
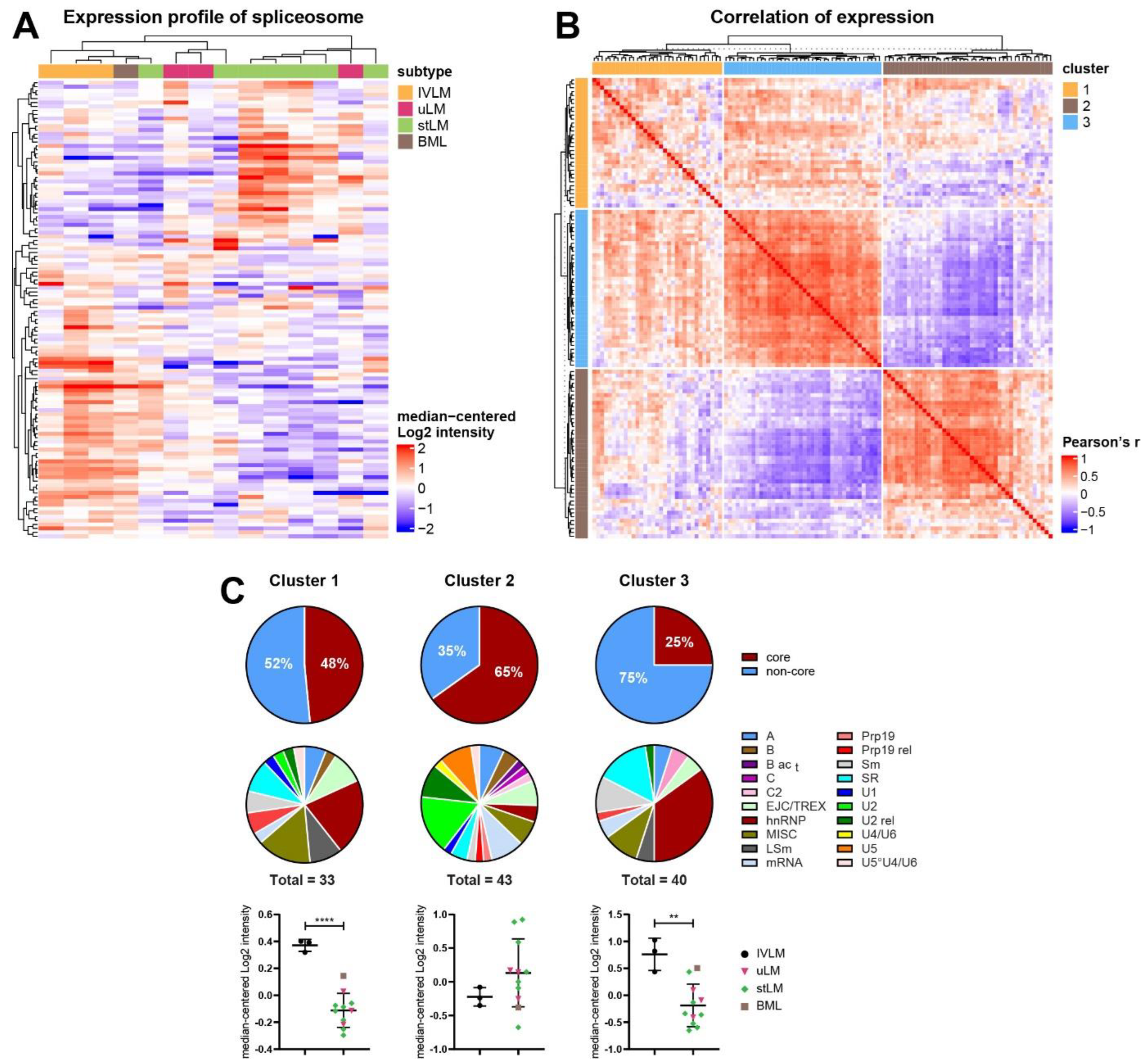
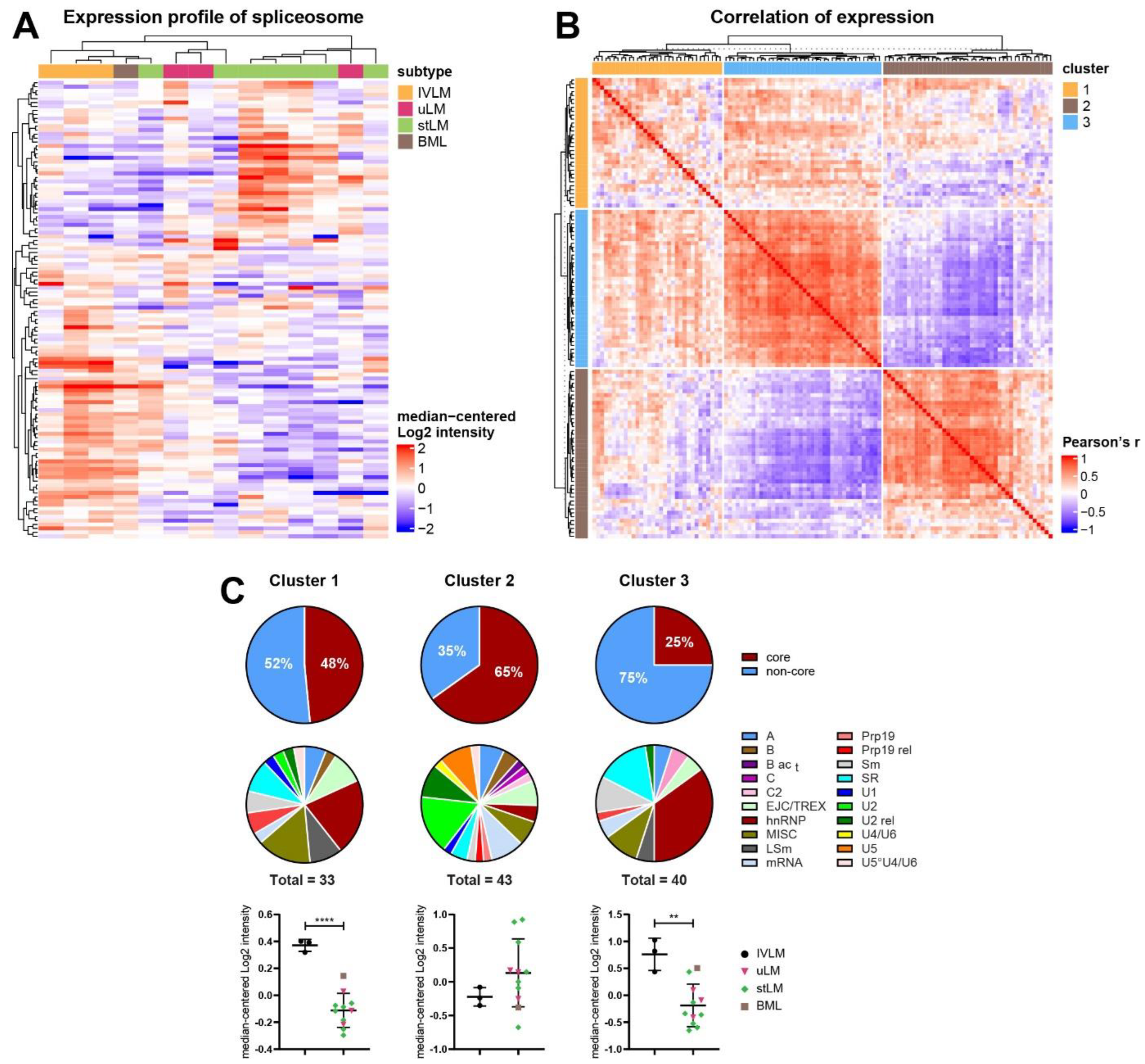
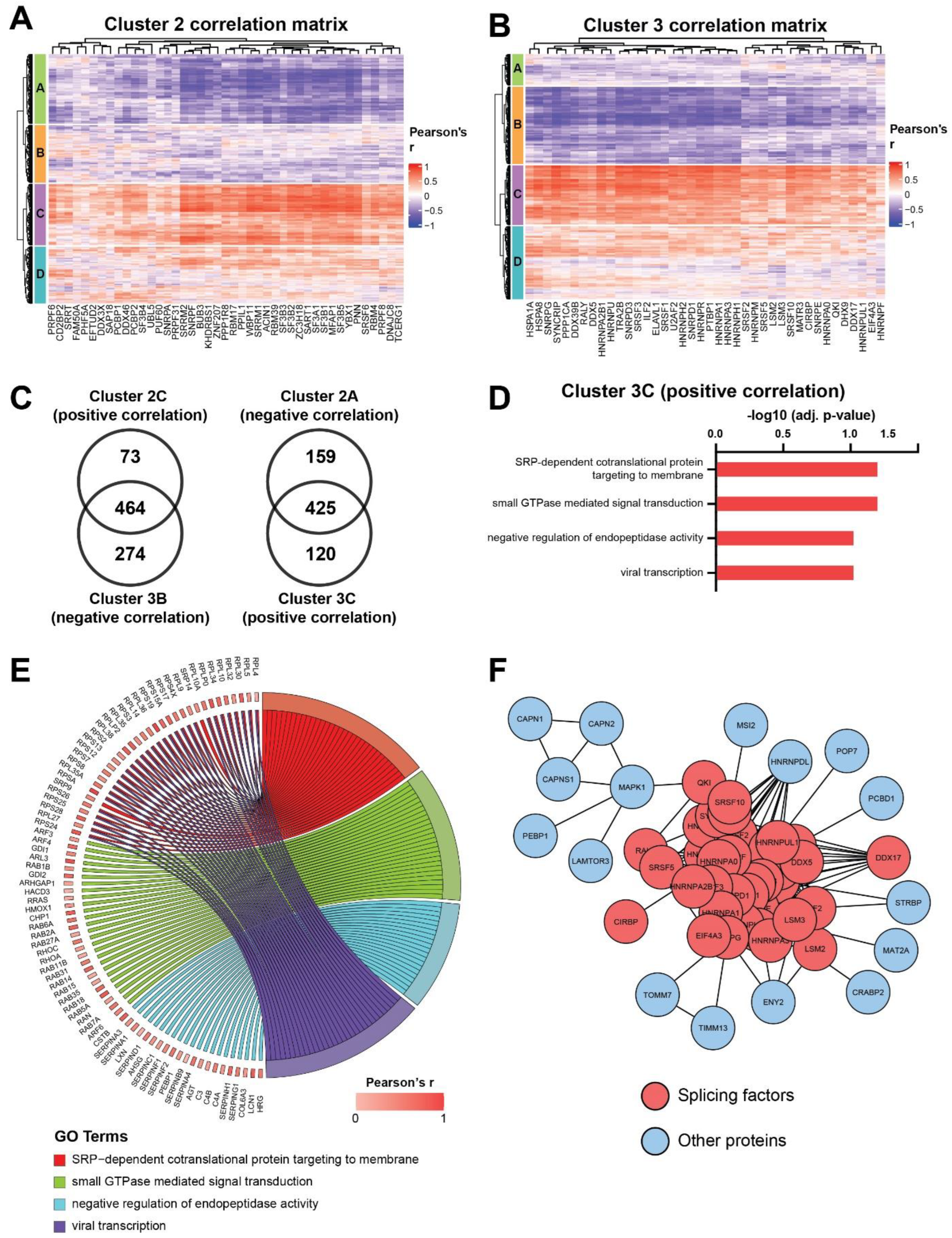
3.3. Identification of Co-Regulated Expression of Splicing Factors in the Proteomic Profiling Dataset
3.4. Distinct Co-Regulated Clusters Are Comprised of Splicing Factors Which Are Differentially Expressed in IVLM versus the Other Smooth Muscle Tumours
3.5. Co-Regulated Splicing Factors Are Associated with Multiple Biological Pathways, including Protein Translocation and Signal Transduction by Small GTPases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valdes Devesa, V.; Conley, C.R.; Stone, W.M.; Collins, J.M.; Magrina, J.F. Update on intravenous leiomyomatosis: Report of five patients and literature review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 171, 209–213. [Google Scholar] [CrossRef]
- Clement, P.B.; Young, R.H.; Scully, R.E. Intravenous leiomyomatosis of the uterus. A clinicopathological analysis of 16 cases with unusual histologic features. Am. J. Surg. Pathol. 1988, 12, 932–945. [Google Scholar] [CrossRef]
- Ling, F.T.; David, T.E.; Merchant, N.; Yu, E.; Butany, J.W. Intracardiac extension of intravenous leiomyomatosis in a pregnant woman: A case report and review of the literature. Can. J. Cardiol. 2000, 16, 73–79. [Google Scholar]
- Lo, K.W.; Lau, T.K. Intracardiac leiomyomatosis. Case report and literature review. Arch. Gynecol. Obstet. 2001, 264, 209–210. [Google Scholar] [CrossRef]
- Konrad, P.; Mellblom, L. Intravenous leiomyomatosis. Acta. Obstet. Gynecol. Scand. 1989, 68, 371–376. [Google Scholar] [CrossRef]
- Carr, R.J.; Hui, P.; Buza, N. Intravenous leiomyomatosis revisited: An experience of 14 cases at a single medical center. Int. J. Gynecol. Pathol. 2015, 34, 169–176. [Google Scholar] [CrossRef]
- Pacheco-Rodriguez, G.; Taveira-DaSilva, A.M.; Moss, J. Benign Metastasizing Leiomyoma. Clin. Chest Med. 2016, 37, 589–595. [Google Scholar] [CrossRef]
- Altinok, G.; Usubutun, A.; Kucukali, T.; Gunalp, S.; Ayhan, A. Disseminated peritoneal leiomyomatosis. A benign entity mimicking carcinomatosis. Arch. Gynecol. Obstet. 2000, 264, 54–55. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Chen, F.; Zhang, M.; Jia, R.; Liu, X.; Zhang, C.; Shao, J.; Cheng, N.; Ma, G.; et al. Intravenous leiomyomatosis is inclined to a solid entity different from uterine leiomyoma based on RNA-seq analysis with RT-qPCR validation. Cancer Med. 2020, 9, 4581–4592. [Google Scholar] [CrossRef]
- Wang, L.; Hu, S.; Xin, F.; Zhao, H.; Li, G.; Ran, W.; Xing, X.; Wang, J. MED12 exon 2 mutation is uncommon in intravenous leiomyomatosis: Clinicopathologic features and molecular study. Hum. Pathol. 2020, 99, 36–42. [Google Scholar] [CrossRef]
- Ordulu, Z.; Chai, H.; Peng, G.; McDonald, A.G.; De Nictolis, M.; Garcia-Fernandez, E.; Hardisson, D.; Prat, J.; Li, P.; Hui, P.; et al. Molecular and clinicopathologic characterization of intravenous leiomyomatosis. Mod. Pathol. 2020, 33, 1844–1860. [Google Scholar] [CrossRef]
- Lu, B.; Liu, Q.; Tang, L.; Ma, Y.; Shi, H. Intravenous leiomyomatosis: Molecular analysis of 17 cases. Pathology 2020, 52, 213–217. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, L.; Xu, R.; Zhu, C.; Ma, G.; Zhang, C.; Liu, X.; Zhao, H.; Miao, Q. Identification of the molecular relationship between intravenous leiomyomatosis and uterine myoma using RNA sequencing. Sci. Rep. 2019, 9, 1442. [Google Scholar] [CrossRef]
- Ordulu, Z.; Nucci, M.R.; Dal Cin, P.; Hollowell, M.L.; Otis, C.N.; Hornick, J.L.; Park, P.J.; Kim, T.M.; Quade, B.J.; Morton, C.C. Intravenous leiomyomatosis: An unusual intermediate between benign and malignant uterine smooth muscle tumors. Mod. Pathol. 2016, 29, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Buza, N.; Xu, F.; Wu, W.; Carr, R.J.; Li, P.; Hui, P. Recurrent chromosomal aberrations in intravenous leiomyomatosis of the uterus: High-resolution array comparative genomic hybridization study. Hum. Pathol. 2014, 45, 1885–1892. [Google Scholar] [CrossRef]
- Burns, J.; Wilding, C.P.; Jones, R.J.; Huang, P.H. Proteomic research in sarcomas—Current status and future opportunities. Semin. Cancer Biol. 2020, 61, 56–70. [Google Scholar] [CrossRef]
- Noujaim, J.; Payne, L.S.; Judson, I.; Jones, R.L.; Huang, P.H. Phosphoproteomics in translational research: A sarcoma perspective. Ann. Oncol. 2016, 27, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Milighetti, M.; Krasny, L.; Lee, A.T.J.; Morani, G.; Szecsei, C.; Chen, Y.; Guljar, N.; McCarthy, F.; Wilding, C.P.; Arthur, A.; et al. Proteomic profiling of soft tissue sarcomas with SWATH mass spectrometry. J. Proteom. 2021, 241, 104236. [Google Scholar] [CrossRef]
- Krasny, L.; Huang, P.H. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol. Omics. 2021, 17, 29–42. [Google Scholar] [CrossRef]
- Judson, I.; Messiou, C. Vitamin D deficiency in the pathogenesis of leiomyoma and intravascular leiomyomatosis: A case report and review of the literature. Gynecol. Oncol. Rep. 2021, 35, 100681. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, G.; Koh, C.C.; Guo, T.; Rost, H.L.; Kouvonen, P.; Collins, B.C.; Heusel, M.; Liu, Y.; Caron, E.; Vichalkovski, A.; et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data 2014, 1, 140031. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Cuklina, J.; Lee, C.H.; Williams, E.G.; Sajic, T.; Collins, B.C.; Rodriguez, M.M.; Sharma, V.S.; Wendt, F.; Goetze, S.; Keele, G.R.; et al. Diagnostics and correction of batch effects in large-scale proteomic studies: A tutorial. Mol. Syst. Biol. 2021, 17, e10240. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Hegele, A.; Kamburov, A.; Grossmann, A.; Sourlis, C.; Wowro, S.; Weimann, M.; Will, C.L.; Pena, V.; Luhrmann, R.; Stelzl, U. Dynamic protein-protein interaction wiring of the human spliceosome. Mol. Cell 2012, 45, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvitkovic, I.; Jurica, M.S. Spliceosome database: A tool for tracking components of the spliceosome. Nucleic Acids Res. 2013, 41, D132–D141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome. Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Morris, J.H.; Apeltsin, L.; Newman, A.M.; Baumbach, J.; Wittkop, T.; Su, G.; Bader, G.D.; Ferrin, T.E. clusterMaker: A multi-algorithm clustering plugin for Cytoscape. BMC Bioinform. 2011, 12, 436. [Google Scholar] [CrossRef] [Green Version]
- Galindo, L.J.; Hernandez-Beeftink, T.; Salas, A.; Jung, Y.; Reyes, R.; de Oca, F.M.; Hernandez, M.; Almeida, T.A. HMGA2 and MED12 alterations frequently co-occur in uterine leiomyomas. Gynecol. Oncol. 2018, 150, 562–568. [Google Scholar] [CrossRef]
- Gattas, G.J.; Quade, B.J.; Nowak, R.A.; Morton, C.C. HMGIC expression in human adult and fetal tissues and in uterine leiomyomata. Genes Chromosomes Cancer 1999, 25, 316–322. [Google Scholar] [CrossRef]
- Gross, K.L.; Neskey, D.M.; Manchanda, N.; Weremowicz, S.; Kleinman, M.S.; Nowak, R.A.; Ligon, A.H.; Rogalla, P.; Drechsler, K.; Bullerdiek, J.; et al. HMGA2 expression in uterine leiomyomata and myometrium: Quantitative analysis and tissue culture studies. Genes Chromosomes Cancer 2003, 38, 68–79. [Google Scholar] [CrossRef]
- Klotzbucher, M.; Wasserfall, A.; Fuhrmann, U. Misexpression of wild-type and truncated isoforms of the high-mobility group I proteins HMGI-C and HMGI(Y) in uterine leiomyomas. Am. J. Pathol. 1999, 155, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koedoot, E.; Smid, M.; Foekens, J.A.; Martens, J.W.M.; Le Devedec, S.E.; van de Water, B. Co-regulated gene expression of splicing factors as drivers of cancer progression. Sci. Rep. 2019, 9, 5484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillette, M.A.; Satpathy, S.; Cao, S.; Dhanasekaran, S.M.; Vasaikar, S.V.; Krug, K.; Petralia, F.; Li, Y.; Liang, W.W.; Reva, B.; et al. Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 2020, 182, 200–225 e235. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Buccitelli, C.; Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Wilkinson, M.E.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research, N.; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224 e216. [Google Scholar] [CrossRef] [Green Version]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef] [PubMed]
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; de la Grange, P.; Roman-Roman, S.; et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013, 3, 1122–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, S.L.; Leonidou, A.; Wai, P.; Marchio, C.; Ng, C.K.; Sapino, A.; Salomon, A.V.; Reis-Filho, J.S.; Weigelt, B.; Natrajan, R.C. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J. Pathol. 2015, 235, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Bruscaggin, A.; Spina, V.; Rasi, S.; Khiabanian, H.; Messina, M.; Fangazio, M.; Vaisitti, T.; Monti, S.; Chiaretti, S.; et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: Association with progression and fludarabine-refractoriness. Blood 2011, 118, 6904–6908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, W.B.; Helmenstine, E.; Walsh, N.; Gondek, L.P.; Kelkar, D.S.; Read, A.; Natrajan, R.; Christenson, E.S.; Roman, B.; Das, S.; et al. Hotspot SF3B1 mutations induce metabolic reprogramming and vulnerability to serine deprivation. J. Clin. Investig. 2019, 129, 4708–4723. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M.; et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 2018, 34, 225–241 e228. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Ahmed, D.; Dolatshad, H.; Tatwavedi, D.; Schulze, U.; Sanchi, A.; Ryley, S.; Dhir, A.; Carpenter, L.; Watt, S.M.; et al. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 2020, 34, 2525–2530. [Google Scholar] [CrossRef]
- Awonuga, A.O.; Shavell, V.I.; Imudia, A.N.; Rotas, M.; Diamond, M.P.; Puscheck, E.E. Pathogenesis of benign metastasizing leiomyoma: A review. Obstet. Gynecol. Surv. 2010, 65, 189–195. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overall | IVLM | LMM | uLMM | BLM | ||
|---|---|---|---|---|---|---|
| Number of cases | 14 | 3 | 7 | 3 | 1 | |
| Age | 41.6 (16–63) | 43 (36–51) | 40.3 (16–63) | 43 (39–50) | 42 | |
| Presenting Symptom | Abdominal/pelvic mass | 6 | 0 | 4 | 2 | 0 |
| Inguinal mass | 3 | 0 | 3 | 0 | 0 | |
| Abnormal vaginal bleeding | 2 | 0 | 0 | 1 | 1 | |
| Other * | 2 | 2 | 0 | 0 | 0 | |
| N/A | 1 | 1 | 0 | 0 | 0 | |
| Anatomical site | Vasculature | 3 | 3 | 0 | 0 | 0 |
| Abdomen | 7 | 0 | 7 | 0 | 0 | |
| Uterus | 4 | 0 | 0 | 3 | 1 | |
| Tumour size (mm) | 71.6 (35–120) | 175 (24–390) | 108 (54–160) | 250 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasny, L.; Wilding, C.P.; Perkins, E.; Arthur, A.; Guljar, N.; Jenks, A.D.; Fisher, C.; Judson, I.; Thway, K.; Jones, R.L.; et al. Proteomic Profiling Identifies Co-Regulated Expression of Splicing Factors as a Characteristic Feature of Intravenous Leiomyomatosis. Cancers 2022, 14, 2907. https://doi.org/10.3390/cancers14122907
Krasny L, Wilding CP, Perkins E, Arthur A, Guljar N, Jenks AD, Fisher C, Judson I, Thway K, Jones RL, et al. Proteomic Profiling Identifies Co-Regulated Expression of Splicing Factors as a Characteristic Feature of Intravenous Leiomyomatosis. Cancers. 2022; 14(12):2907. https://doi.org/10.3390/cancers14122907
Chicago/Turabian StyleKrasny, Lukas, Chris P. Wilding, Emma Perkins, Amani Arthur, Nafia Guljar, Andrew D. Jenks, Cyril Fisher, Ian Judson, Khin Thway, Robin L. Jones, and et al. 2022. "Proteomic Profiling Identifies Co-Regulated Expression of Splicing Factors as a Characteristic Feature of Intravenous Leiomyomatosis" Cancers 14, no. 12: 2907. https://doi.org/10.3390/cancers14122907
APA StyleKrasny, L., Wilding, C. P., Perkins, E., Arthur, A., Guljar, N., Jenks, A. D., Fisher, C., Judson, I., Thway, K., Jones, R. L., & Huang, P. H. (2022). Proteomic Profiling Identifies Co-Regulated Expression of Splicing Factors as a Characteristic Feature of Intravenous Leiomyomatosis. Cancers, 14(12), 2907. https://doi.org/10.3390/cancers14122907