
Multiple Components of Protein Homeostasis Pathway Can Be Targeted to Produce Drug Synergies with VCP Inhibitors in Ovarian Cancer


Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines and Cell Culture
2.3. Cell Viability Assay and Drug Synergy Studies
2.4. Clonogenic Assay and 3D Spheroid Assay
2.5. Transient Small Interfering RNA (siRNA) Knockdown and Plasmid Transfection
2.6. Caspase-3 Activity Assay
2.7. Western Blot and Antibodies
3. Results
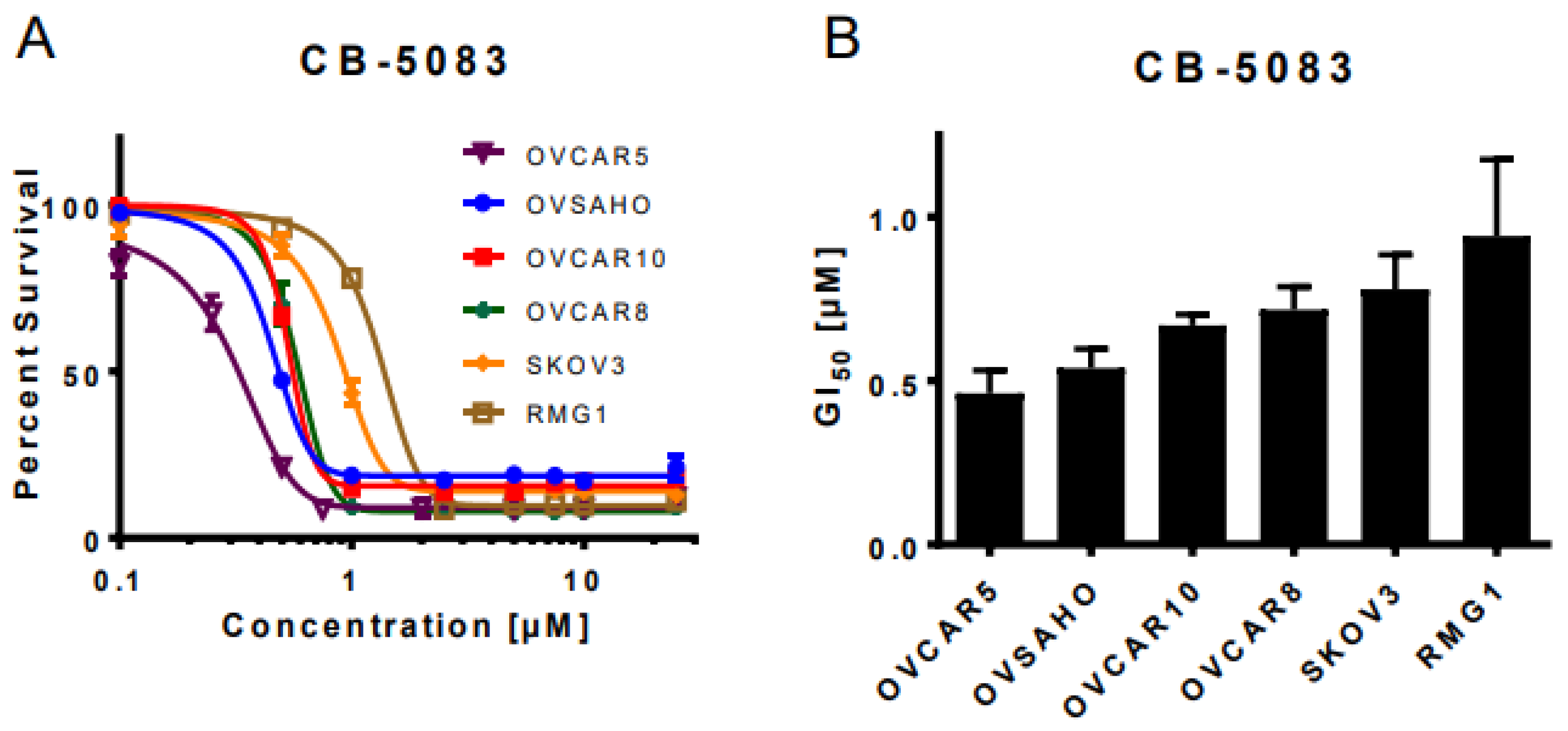
3.1. VCP Inhibitor CB-5083 Treatment Induces Cytotoxicity in Ovarian Cancer Cells
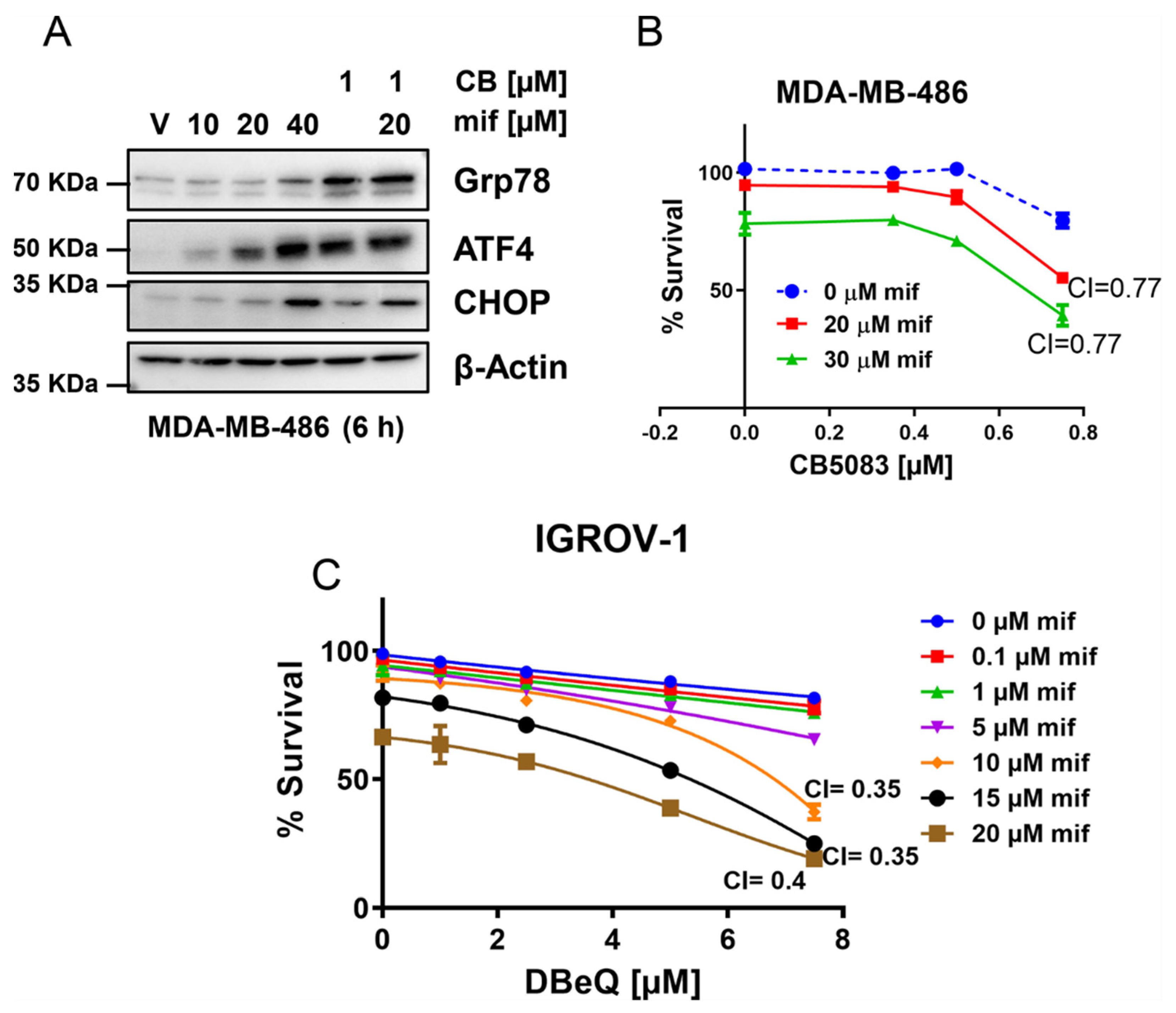
3.2. VCP Inhibitors Show Synergistic Cytotoxicity with Clinically Achievable Doses of Mifepristone
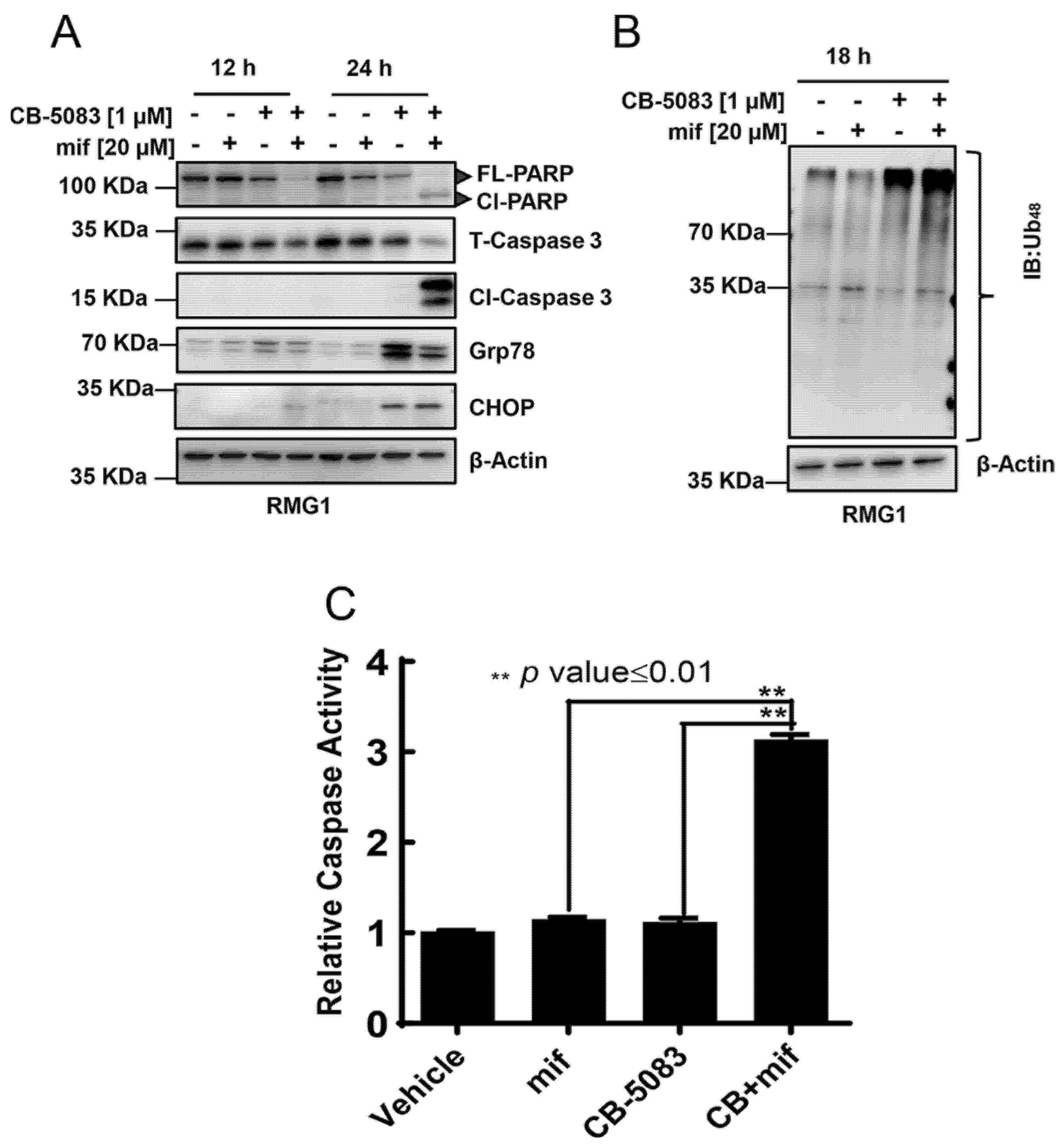
3.3. CB-5083 and Mifepristone Combination Enhances Caspase Activity and Cytotoxicity
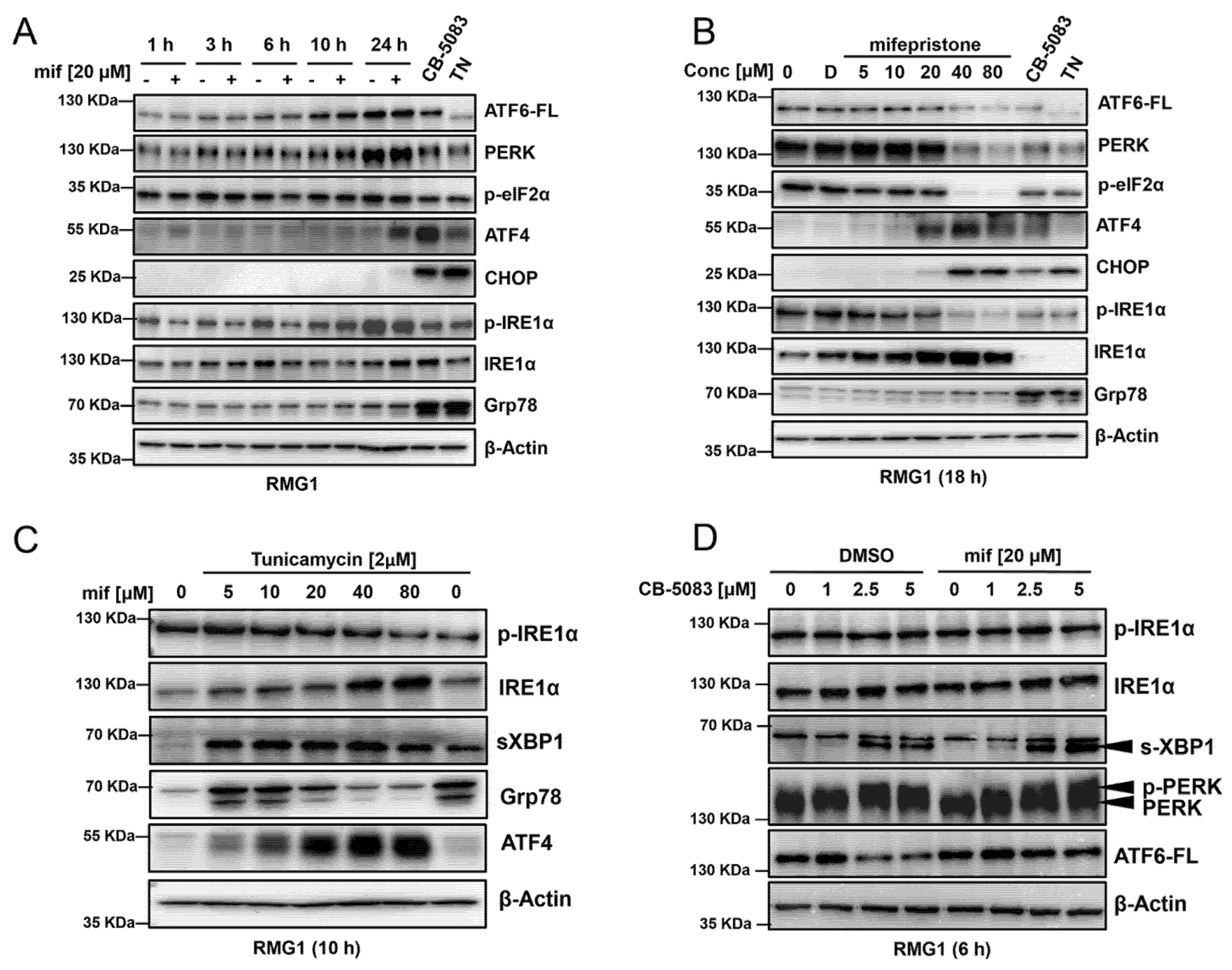
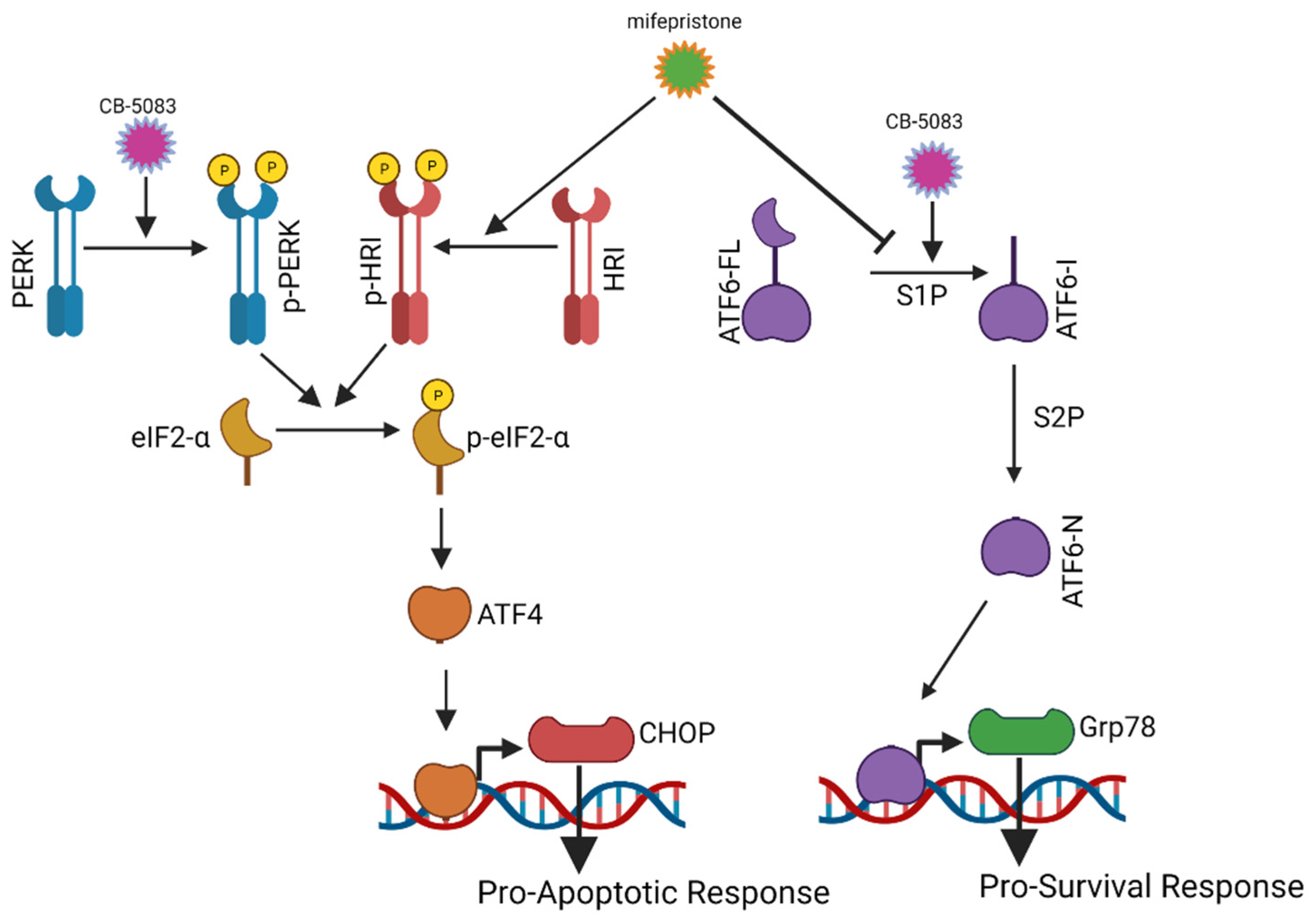
3.4. Mifepristone Activates the Unfolded Protein Response Independent of Glucocorticoid, Estrogen, and Progesterone Receptor Inhibition
3.5. Mifepristone Blocks ATF6 Signaling
3.6. Mifepristone Activates Heme-Regulated Inhibitor (HRI) Pathway to Induce Activating Transcription Factor 4 (ATF4)
3.7. Targeting Multiple Components of Proteostasis Produces Synergistic Drug Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Berkenblit, A.; Cannistra, S.A. Advances in the management of epithelial ovarian cancer. J. Reprod. Med. 2005, 50, 426–438. [Google Scholar] [PubMed]
- Armstrong, D.K. Relapsed ovarian cancer: Challenges and management strategies for a chronic disease. Oncologist 2002, 7 (Suppl. 5), 20–28. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, S.; Smeets, D.; Moisse, M.; Braicu, E.I.; Vanderstichele, A.; Zhao, H.; Van Nieuwenhuysen, E.; Berns, E.; Sehouli, J.; Zeillinger, R.; et al. Genetic heterogeneity after first-line chemotherapy in high-grade serous ovarian cancer. Eur. J. Cancer 2016, 53, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Aoki, D.; Chiyoda, T. PARP inhibitors and quality of life in ovarian cancer. Lancet Oncol. 2018, 19, 1012–1014. [Google Scholar] [CrossRef]
- Marcotte, R.; Brown, K.R.; Suarez, F.; Sayad, A.; Karamboulas, K.; Krzyzanowski, P.M.; Sircoulomb, F.; Medrano, M.; Fedyshyn, Y.; Koh, J.L.; et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012, 2, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Cheung, H.W.; Cowley, G.S.; Weir, B.A.; Boehm, J.S.; Rusin, S.; Scott, J.A.; East, A.; Ali, L.D.; Lizotte, P.H.; Wong, T.C.; et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 12372–12377. [Google Scholar] [CrossRef] [Green Version]
- Etemadmoghadam, D.; Weir, B.A.; Au-Yeung, G.; Alsop, K.; Mitchell, G.; George, J.; Australian Ovarian Cancer Study, G.; Davis, S.; D’Andrea, A.D.; Simpson, K.; et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc. Natl. Acad. Sci. USA 2013, 110, 19489–19494. [Google Scholar] [CrossRef] [Green Version]
- Bastola, P.; Oien, D.B.; Cooley, M.; Chien, J. Emerging Cancer Therapeutic Targets in Protein Homeostasis. AAPS J. 2018, 20, 94. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [Green Version]
- Queitsch, C.; Sangster, T.A.; Lindquist, S. Hsp90 as a capacitor of phenotypic variation. Nature 2002, 417, 618–624. [Google Scholar] [CrossRef]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar] [CrossRef]
- Bastola, P.; Neums, L.; Schoenen, F.J.; Chien, J. VCP inhibitors induce endoplasmic reticulum stress, cause cell cycle arrest, trigger caspase-mediated cell death and synergistically kill ovarian cancer cells in combination with Salubrinal. Mol. Oncol. 2016, 10, 1559–1574. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.J.; Le Moigne, R.; Djakovic, S.; Kumar, B.; Rice, J.; Wong, S.; Wang, J.; Yao, B.; Valle, E.; Kiss von Soly, S.; et al. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell 2015, 28, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, H.; Cheng, C.; Pitkanen, M.; Sander, C.L.; Zhang, J.; Saeid, S.; Turunen, T.; Shmara, A.; Weiss, L.; Ta, L.; et al. A p97/Valosin-Containing Protein Inhibitor Drug CB-5083 Has a Potent but Reversible Off-Target Effect on Phosphodiesterase-6. J. Pharmacol. Exp. Ther. 2021, 378, 31–41. [Google Scholar] [CrossRef]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [Green Version]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J.; et al. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012, 2, e79. [Google Scholar] [CrossRef]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [Green Version]
- Baulieu, E.E. Contragestion and other clinical applications of RU 486, an antiprogesterone at the receptor. Science 1989, 245, 1351–1357. [Google Scholar] [CrossRef]
- Kretschmer, X.C.; Baldwin, W.S. CAR and PXR: Xenosensors of endocrine disrupters? Chem. Biol. Interact. 2005, 155, 111–128. [Google Scholar] [CrossRef]
- Grunberg, S.M.; Weiss, M.H.; Russell, C.A.; Spitz, I.M.; Ahmadi, J.; Sadun, A.; Sitruk-Ware, R. Long-term administration of mifepristone (RU486): Clinical tolerance during extended treatment of meningioma. Cancer Investig. 2006, 24, 727–733. [Google Scholar] [CrossRef]
- Liu, R.; Shi, P.; Nie, Z.; Liang, H.; Zhou, Z.; Chen, W.; Chen, H.; Dong, C.; Yang, R.; Liu, S.; et al. Mifepristone Suppresses Basal Triple-Negative Breast Cancer Stem Cells by Down-regulating KLF5 Expression. Theranostics 2016, 6, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Goyeneche, A.A.; Caron, R.W.; Telleria, C.M. Mifepristone inhibits ovarian cancer cell growth in vitro and in vivo. Clin. Cancer Res. 2007, 13, 3370–3379. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hapon, M.B.; Goyeneche, A.A.; Srinivasan, R.; Gamarra-Luques, C.D.; Callegari, E.A.; Drappeau, D.D.; Terpstra, E.J.; Pan, B.; Knapp, J.R.; et al. Mifepristone increases mRNA translation rate, triggers the unfolded protein response, increases autophagic flux, and kills ovarian cancer cells in combination with proteasome or lysosome inhibitors. Mol. Oncol. 2016, 10, 1099–1117. [Google Scholar] [CrossRef] [Green Version]
- Dioufa, N.; Kassi, E.; Papavassiliou, A.G.; Kiaris, H. Atypical induction of the unfolded protein response by mifepristone. Endocrine 2010, 38, 167–173. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [Green Version]
- Bastola, P.; Minn, K.; Chien, J. Heterozygous mutations in p97 and resistance to p97 inhibitors. bioRxiv 2018. [Google Scholar] [CrossRef]
- Chen, H.; Gotimer, K.; De Souza, C.; Tepper, C.G.; Karnezis, A.N.; Leiserowitz, G.S.; Chien, J.; Smith, L.H. Short-term organoid culture for drug sensitivity testing of high-grade serous carcinoma. Gynecol. Oncol. 2020, 157, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Bastola, P.; Wang, F.; Schaich, M.A.; Gan, T.; Freudenthal, B.D.; Chou, T.F.; Chien, J. Specific mutations in the D1-D2 linker region of VCP/p97 enhance ATPase activity and confer resistance to VCP inhibitors. Cell Death Discov. 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Chien, J.; Mehta, G. 11th Biennial Ovarian Cancer Research Symposium. In Proceedings of the Ovarian Cancer Research Symposium at the Rivkin Center for Ovarian Cancer, Seattle, WA, USA, 27 November 2017. [Google Scholar]
- Agarwai, M.K. The antiglucocorticoid action of mifepristone. Pharmacol. Ther. 1996, 70, 183–213. [Google Scholar] [CrossRef]
- Subik, K.; Lee, J.F.; Baxter, L.; Strzepek, T.; Costello, D.; Crowley, P.; Xing, L.; Hung, M.C.; Bonfiglio, T.; Hicks, D.G.; et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer 2010, 4, 35–41. [Google Scholar] [CrossRef]
- Stringer-Reasor, E.M.; Baker, G.M.; Skor, M.N.; Kocherginsky, M.; Lengyel, E.; Fleming, G.F.; Conzen, S.D. Glucocorticoid Receptor Activation Inhibits Chemotherapy-induced Cell Death in High-grade Serous Ovarian Carcinoma. Gynecol. Oncol. 2015, 138, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Baumeister, P.; Luo, S.; Skarnes, W.C.; Sui, G.; Seto, E.; Shi, Y.; Lee, A.S. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: Activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol. Cell. Biol. 2005, 25, 4529–4540. [Google Scholar] [CrossRef] [Green Version]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021, 22, 343. [Google Scholar] [CrossRef]
- Blayney, J.K.; Davison, T.; McCabe, N.; Walker, S.; Keating, K.; Delaney, T.; Greenan, C.; Williams, A.R.; McCluggage, W.G.; Capes-Davis, A.; et al. Prior knowledge transfer across transcriptional data sets and technologies using compositional statistics yields new mislabelled ovarian cell line. Nucleic Acids Res. 2016, 44, e137. [Google Scholar] [CrossRef] [Green Version]
- Naoki, T.; Ikuya, S.; Osamu, T.; Keishi, M. RU486, a progestin antagonist, binds to progesterone receptors in a human endometrial cancer cell line and reverses the growth inhibition by progestins. J. Steroid Biochem. 1988, 31, 161–166. [Google Scholar] [CrossRef]
- Gallagher, C.M.; Garri, C.; Cain, E.L.; Ang, K.K.-H.; Wilson, C.G.; Chen, S.; Hearn, B.R.; Jaishankar, P.; Aranda-Diaz, A.; Arkin, M.R.; et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. eLife 2016, 5, e11878. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Gyorffy, B.; Lanczky, A.; Szallasi, Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr. Relat. Cancer 2012, 19, 197–208. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastola, P.; Leiserowitz, G.S.; Chien, J. Multiple Components of Protein Homeostasis Pathway Can Be Targeted to Produce Drug Synergies with VCP Inhibitors in Ovarian Cancer. Cancers 2022, 14, 2949. https://doi.org/10.3390/cancers14122949
Bastola P, Leiserowitz GS, Chien J. Multiple Components of Protein Homeostasis Pathway Can Be Targeted to Produce Drug Synergies with VCP Inhibitors in Ovarian Cancer. Cancers. 2022; 14(12):2949. https://doi.org/10.3390/cancers14122949
Chicago/Turabian StyleBastola, Prabhakar, Gary S. Leiserowitz, and Jeremy Chien. 2022. "Multiple Components of Protein Homeostasis Pathway Can Be Targeted to Produce Drug Synergies with VCP Inhibitors in Ovarian Cancer" Cancers 14, no. 12: 2949. https://doi.org/10.3390/cancers14122949