
The Homologous Recombination Deficiency Scar in Advanced Cancer: Agnostic Targeting of Damaged DNA Repair


 ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
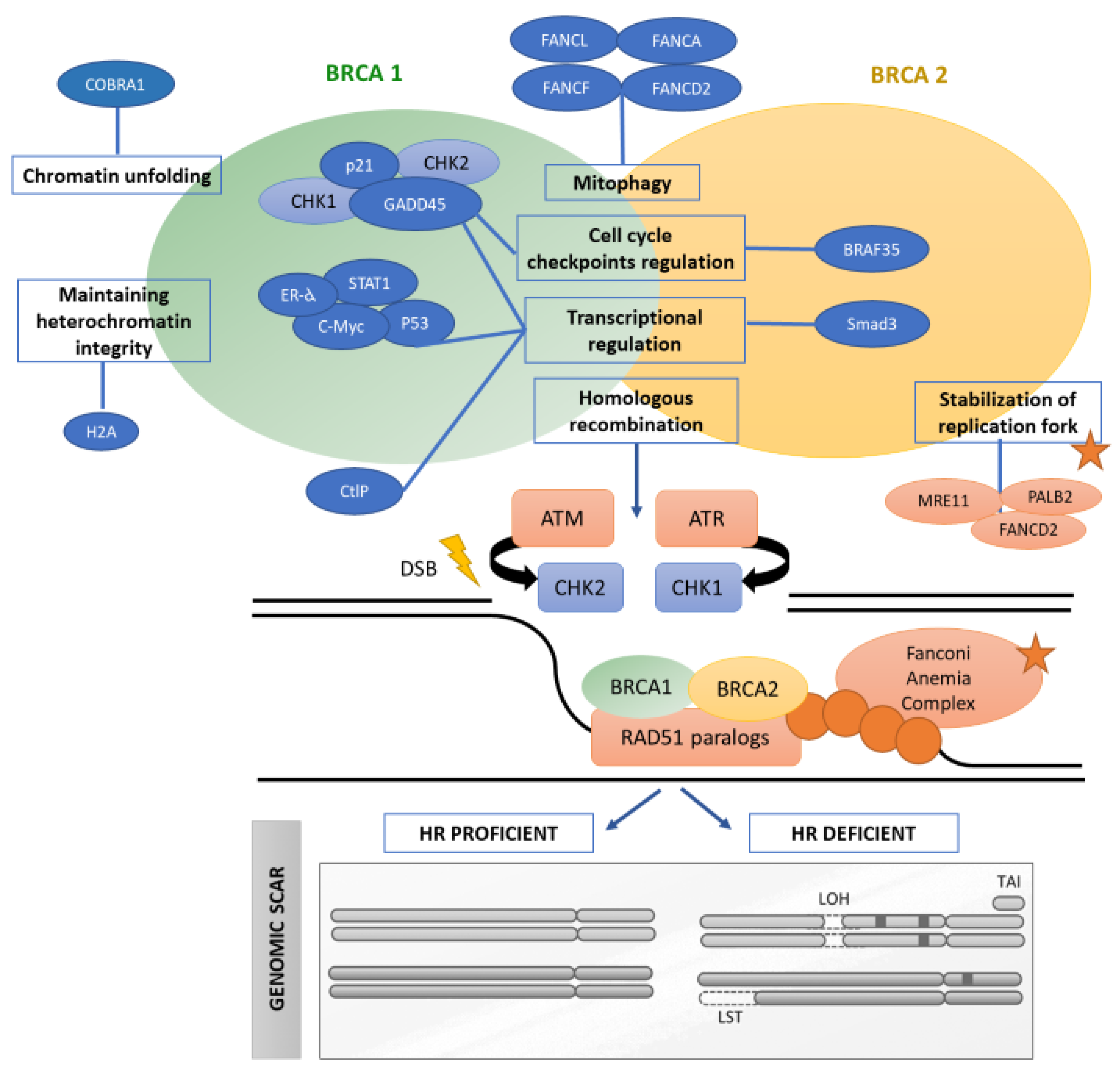
2. Homologous Recombination: A Key Pathway in DNA Repair
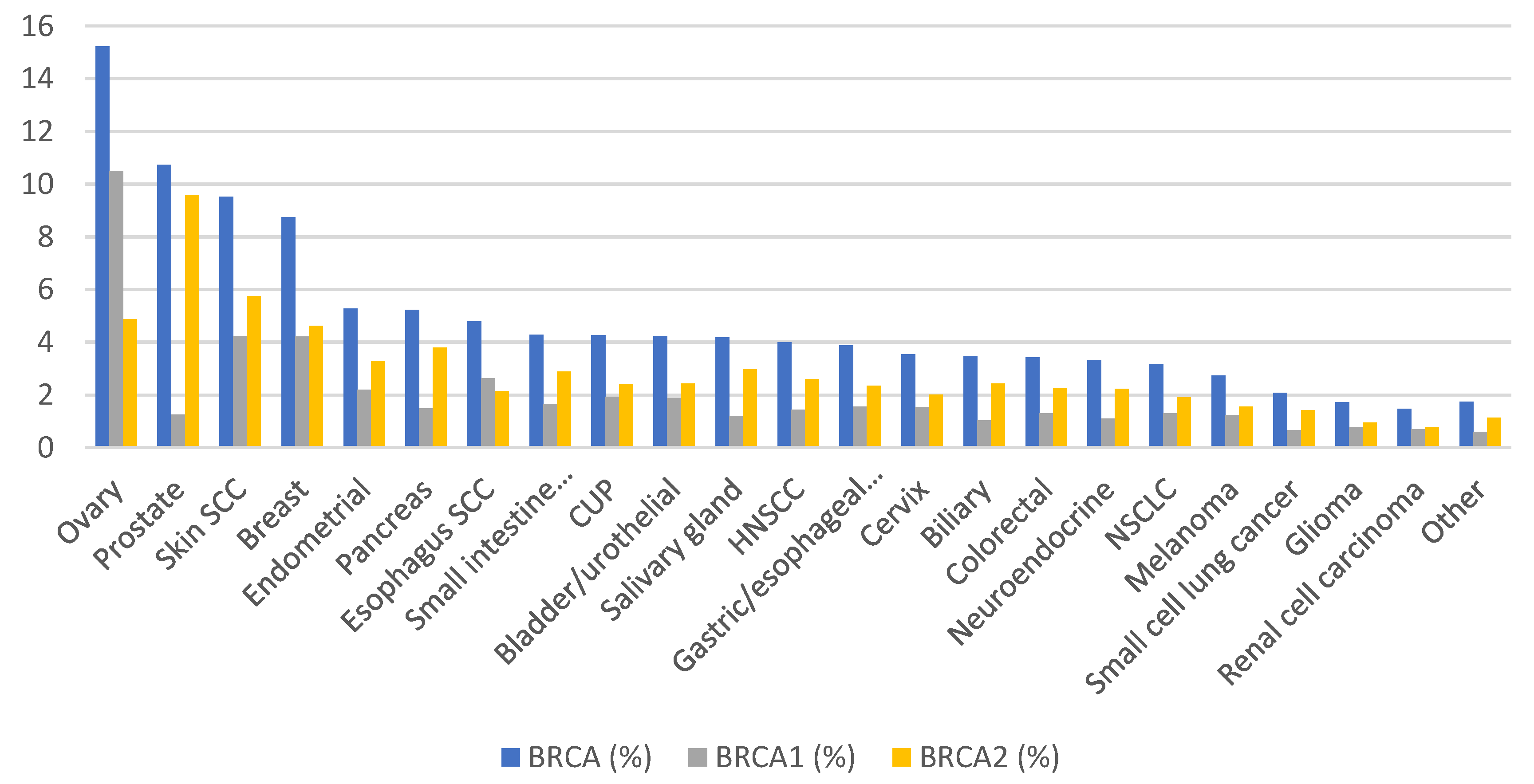
3. Prevalence and Prognostic Value of HRD in Cancer
4. HRD as an Actionable Target
4.1. Platinum
4.2. PARP Inhibitors
5. The Challenge of Testing: Searching for HRD Scar
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Vega, D.M.; Arend, R.C.; Baden, J.F.; Barbash, O.; Beaubier, N.; Collins, G.; French, T.; Ghahramani, N.; Hinson, P.; et al. Homologous Recombination Deficiency: Concepts, Definitions, and Assays. Oncologist 2022, 27, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. JNCI J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA genes: The role in genome stability, cancer stemness and therapy resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of ATM-Dependent Phosphorylation of Brca1 in the DNA Damage Response to Double-Strand Breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef]
- Tibbetts, R.S.; Cortez, D.; Brumbaugh, K.M.; Scully, R.; Livingston, D.; Elledge, S.J.; Abraham, R.T. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000, 14, 2989–3002. [Google Scholar] [CrossRef]
- Lee, J.-S.; Collins, K.M.; Brown, A.L.; Lee, C.-H.; Chung, J.H. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 2000, 404, 201–204. [Google Scholar] [CrossRef]
- Chen, J. Ataxia telangiectasia-related protein is involved in the phosphorylation of BRCA1 following deoxyribonucleic acid damage. Cancer Res. 2000, 60, 5037–5039. [Google Scholar] [PubMed]
- Deng, C.-X. BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef]
- Badie, S.; Escandell, J.M.; Bouwman, P.; Carlos, A.R.; Thanasoula, M.; Gallardo, M.M.; Suram, A.; Jaco, I.; Benitez, J.; Herbig, U.; et al. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat. Struct. Mol. Biol. 2010, 17, 1461–1469. [Google Scholar] [CrossRef]
- Hasty, P.; Montagna, C. Chromosomal rearrangements in cancer: Detection and potential causal mechanisms. Mol. Cell. Oncol. 2014, 1, e29904. [Google Scholar] [CrossRef]
- Lotan, T.L.; Kaur, H.B.; Salles, D.C.; Murali, S.; Schaeffer, E.M.; Lanchbury, J.S.; Isaacs, W.B.; Brown, R.; Richardson, A.L.; Cussenot, O.; et al. Homologous recombination deficiency (HRD) score in germline BRCA2- versus ATM-altered prostate cancer. Mod. Pathol. 2021, 34, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Norquist, B.M.; Brady, M.F.; Harrell, M.I.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Burger, R.A.; Tewari, K.S.; et al. Mutations in Homologous Recombination Genes and Outcomes in Ovarian Carcinoma Patients in GOG 218: An NRG Oncology/Gynecologic Oncology Group Study. Clin. Cancer Res. 2017, 24, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef]
- Sokol, E.S.; Pavlick, D.; Khiabanian, H.; Frampton, G.M.; Ross, J.S.; Gregg, J.P.; Lara, P.N.; Oesterreich, S.; Agarwal, N.; Necchi, A.; et al. Pan-Cancer Analysis of BRCA1 and BRCA2 Genomic Alterations and Their Association with Genomic Instability as Measured by Genome-Wide Loss of Heterozygosity. JCO Precis. Oncol. 2020, 4, 442–465. [Google Scholar] [CrossRef] [PubMed]
- Marquard, A.M.; Eklund, A.C.; Joshi, T.; Krzystanek, M.; Favero, F.; Wang, Z.C.; Richardson, A.L.; Silver, D.P.; Szallasi, Z.; Birkbak, N.J. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark. Res. 2015, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.-Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, D.; Cramer, D.W.; Dao, F.; Dhir, R.; Disaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609. [Google Scholar]
- de Jonge, M.M.; Auguste, A.; van Wijk, L.M.; Schouten, P.C.; Meijers, M.; Ter Haar, N.T.; Smit, V.T.; Nout, R.A.; Glaire, M.A.; Church, D.N.; et al. Frequent homologous recombination deficiency in high-grade endometrial carcinomas. Clin. Cancer Res. 2019, 25, 1087–1097. [Google Scholar] [CrossRef]
- Alsop, K.; Fereday, S.; Meldrum, C.; DeFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation–Positive Women with Ovarian Cancer: A Report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Candido-dos-Reis, F.J.; Song, H.; Goode, E.L.; Cunningham, J.M.; Fridley, B.L.; Larson, M.C.; Alsop, K.; Dicks, E.; Harrington, P.; Ramus, S.J.; et al. Germline mutation in BRCA1 or BRCA2 and ten-year survival for women diagnosed with epithelial ovarian cancer. Clin. Cancer Res. 2015, 21, 652–657. [Google Scholar] [CrossRef]
- Foulkes, W.; Wong, N.; Brunet, J.S.; Bégin, L.R.; Zhang, J.C.; Martinez, J.J.; Rozen, F.; Tonin, P.N.; Narod, S.A.; Karp, S.E.; et al. Germ-line BRCA1 mutation is an adverse prognostic factor in Ashkenazi Jewish women with breast cancer. Clin. Cancer Res. 1997, 3, 2465–2469. [Google Scholar] [PubMed]
- Moller, P.; Evans, D.G.; Reis, M.M.; Gregory, H.; Anderson, E.; Maehle, L.; Lalloo, F.; Howell, A.; Apold, J.; Clark, N.; et al. Surveillance for familial breast cancer: Differences in outcome according toBRCA mutation status. Int. J. Cancer 2007, 121, 1017–1020. [Google Scholar] [CrossRef]
- Nilsson, M.P.; Hartman, L.; Idvall, I.; Kristoffersson, U.; Johannsson, O.T.; Loman, N. Long-term prognosis of early-onset breast cancer in a population-based cohort with a known BRCA1/2 mutation status. Breast Cancer Res. Treat. 2014, 144, 133–142. [Google Scholar] [CrossRef][Green Version]
- Bayraktar, S.; Gutierrez-Barrera, A.M.; Lin, H.; Elsayegh, N.; Tasbas, T.; Litton, J.K.; Ibrahim, N.K.; Morrow, P.K.; Green, M.; Valero, V.; et al. Outcome of metastatic breast cancer in selected women with or without deleterious BRCA mutations. Clin. Exp. Metastasis 2013, 30, 631–642. [Google Scholar] [CrossRef]
- El-Tamer, M.; Russo, D.; Troxel, A.; Bernardino, L.P.; Mazziotta, R.; Estabrook, A.; Ditkoff, B.-A.; Schnabel, F.; Mansukhani, M. Survival and recurrence after breast cancer in BRCA1/2 mutation carriers. Ann. Surg. Oncol. 2004, 11, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Bonadona, V.; Dussart-Moser, S.; Voirin, N.; Sinilnikova, O.M.; Mignotte, H.; Mathevet, P.; Brémond, A.; Treilleux, I.; Martin, A.; Romestaing, P.; et al. Prognosis of early-onset breast cancer based on BRCA1/2 mutation status in a French population-based cohort and review. Breast Cancer Res. Treat. 2007, 101, 233–245. [Google Scholar] [CrossRef]
- Arun, B.; Bayraktar, S.; Liu, D.D.; Barrera, A.M.G.; Atchley, D.; Pusztai, L.; Litton, J.K.; Valero, V.; Meric-Bernstam, F.; Hortobagyi, G.N.; et al. Response to Neoadjuvant Systemic Therapy for Breast Cancer in BRCA Mutation Carriers and Noncarriers: A Single-Institution Experience. J. Clin. Oncol. 2011, 29, 3739–3746. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Phillips, K.-A.; West, D.W.; Ennis, M.; Hopper, J.L.; John, E.M.; O’Malley, F.P.; Milne, R.L.; Andrulis, I.L.; Friedlander, M.L.; et al. Breast Cancer Prognosis in BRCA1 and BRCA2 Mutation Carriers: An International Prospective Breast Cancer Family Registry Population-Based Cohort Study. J. Clin. Oncol. 2012, 30, 19–26. [Google Scholar] [CrossRef]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average Risks of Breast and Ovarian Cancer Associated with BRCA1 or BRCA2 Mutations Detected in Case Series Unselected for Family History: A Combined Analysis of 22 Studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef]
- Armes, J.E.; Egan, A.J.; Southey, M.C.; Dite, G.S.; McCredie, M.R.; Giles, G.; Hopper, J.L.; Venter, D.J. The histologic phenotypes of breast carcinoma occurring before age 40 years in women with and without BRCA1 or BRCA2 germline mutations: A population-based study. Cancer 1998, 83, 2335–2345. [Google Scholar] [CrossRef]
- Southey, M.C.; Ramus, S.; Dowty, J.; Smith, L.D.; Tesoriero, A.A.; Wong, E.M.; Dite, G.; Jenkins, M.; Byrnes, G.; Winship, I.; et al. Morphological predictors of BRCA1 germline mutations in young women with breast cancer. Br. J. Cancer 2011, 104, 903–909. [Google Scholar] [CrossRef]
- Golan, T.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R.; et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Castro, E.; Aragón, I.M.; Cendón, Y.; Cattrini, C.; López-Casas, P.P.; Olmos, D. Genetic aberrations in DNA repair pathways: A cornerstone of precision oncology in prostate cancer. Br. J. Cancer 2020, 124, 552–563. [Google Scholar] [CrossRef]
- Castro, E.; Romero-Laorden, N.; Del Pozo, A.; Lozano, R.; Medina, A.; Puente, J.; Piulats, J.M.; Lorente, D.; Saez, M.I.; Morales-Barrera, R.; et al. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients with Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Paller, C.J.; Antonarakis, E.S.; Beer, T.M.; Borno, H.T.; Carlo, M.I.; George, D.J.; Graff, J.N.; Gupta, S.; Heath, E.I.; Higano, C.S.; et al. Germline Genetic Testing in Advanced Prostate Cancer; Practices and Barriers: Survey Results from the Germline Genetics Working Group of the Prostate Cancer Clinical Trials Consortium. Clin. Genitourin. Cancer 2019, 17, 275–282.e1. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Arun, B.; Hacker, M.R.; Hofstatter, E.; Toppmeyer, D.L.; Isakoff, S.J.; Borges, V.; Legare, R.D.; Isaacs, C.; Wolff, A.C.; et al. TBCRC 031: Randomized Phase II Study of Neoadjuvant Cisplatin Versus Doxorubicin-Cyclophosphamide in Germline BRCA Carriers with HER2-Negative Breast Cancer (the INFORM trial). J. Clin. Oncol. 2020, 38, 1539–1548. [Google Scholar] [CrossRef]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Hahnen, E.; Lederer, B.; Hauke, J.; Loibl, S.; Kröber, S.; Schneeweiss, A.; Denkert, C.; Fasching, P.A.; Blohmer, J.U.; Jackisch, C.; et al. Germline mutation status, pathological complete response, and disease-free survival in triple-negative breast cancer: Secondary analysis of the GeparSixto randomized clinical trial. JAMA Oncol. 2017, 3, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Barenboim, A.; Lahat, G.; Nachmany, I.; Goykhman, Y.; Shacham-Shmueli, E.; Halpern, N.; Brazowski, E.; Geva, R.; Wolf, I.; et al. Increased Rate of Complete Pathologic Response After Neoadjuvant FOLFIRINOX for BRCA Mutation Carriers with Borderline Resectable Pancreatic Cancer. Ann. Surg. Oncol. 2020, 27, 3963–3970. [Google Scholar] [CrossRef]
- Okano, N.; Morizane, C.; Nomura, S.; Takahashi, H.; Tsumura, H.; Satake, H.; Mizuno, N.; Tsuji, K.; Shioji, K.; Asagi, A.; et al. Phase II clinical trial of gemcitabine plus oxaliplatin in patients with metastatic pancreatic adenocarcinoma with a family history of pancreatic/breast/ovarian/prostate cancer or personal history of breast/ovarian/prostate cancer (FABRIC study). Int. J. Clin. Oncol. 2020, 25, 1835–1843. [Google Scholar] [CrossRef]
- Wattenberg, M.; Asch, D.; Yu, S.; O’Dwyer, P.J.; Domchek, S.M.; Nathanson, K.L.; Rosen, M.A.; Beatty, G.L.; Siegelman, E.S.; Reiss, K.A. Platinum response characteristics of patients with pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or PALB2 mutation. Br. J. Cancer 2019, 122, 333–339. [Google Scholar] [CrossRef]
- Schmid, S.; Omlin, A.; Higano, C.; Sweeney, C.; Chanza, N.M.; Mehra, N.; Kuppen, M.C.P.; Beltran, H.; Conteduca, V.; De Almeida, D.V.P.; et al. Activity of Platinum-Based Chemotherapy in Patients with Advanced Prostate Cancer with and without DNA Repair Gene Aberrations. JAMA Netw. Open 2020, 3, e2021692. [Google Scholar] [CrossRef] [PubMed]
- Mota, J.M.; Barnett, E.; Nauseef, J.T.; Nguyen, B.; Stopsack, K.H.; Wibmer, A.; Flynn, J.R.; Heller, G.; Danila, D.C.; Rathkopf, D.; et al. Platinum-Based Chemotherapy in Metastatic Prostate Cancer with DNA Repair Gene Alterations. JCO Precis. Oncol. 2020, 4, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-J.; Xu, Y.; Lin, Y.; Zhu, H.-J.; Zhou, Y.-D.; Mao, F.; Zhang, X.-H.; Huang, X.; Zhong, Y.; Sun, Q.; et al. Platinum-Based Neoadjuvant Chemotherapy for Breast Cancer with BRCA Mutations: A Meta-Analysis. Front. Oncol. 2020, 10, 592998. [Google Scholar] [CrossRef]
- Chai, Y.; Chen Chen, Y.; Zhang, D.; Wei, Y.; Li, Z.; Li, Q.; Xu, B. Homologous Recombination Deficiency (HRD) and BRCA 1/2 Gene Mutation for Predicting the Effect of Plat-inum-Based Neoadjuvant Chemotherapy of Early-Stage Triple-Negative Breast Cancer (TNBC): A Systematic Review and Meta-Analysis. J. Pers. Med. 2022, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Weber, K.E.; Timms, K.M.; Elkin, E.P.; Hahnen, E.; Fasching, P.A.; Lederer, B.; Denkert, C.; Schneeweiss, A.; Braun, S.; et al. Survival analysis of carboplatin added to an anthracycline/taxane-based neoadjuvant chemotherapy and HRD score as predictor of response—Final results from GeparSixto. Ann. Oncol. 2018, 29, 2341–2347. [Google Scholar] [CrossRef]
- Lowery, M.A.; Kelsen, D.P.; Stadler, Z.K.; Yu, K.H.; Janjigian, Y.Y.; Ludwig, E.; D’Adamo, D.R.; Salo-Mullen, E.; Robson, M.E.; Allen, P.J.; et al. An Emerging Entity: Pancreatic Adenocarcinoma Associated with a Known BRCA Mutation: Clinical Descriptors, Treatment Implications, and Future Directions. Oncologist 2011, 16, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Fogelman, D.; Sugar, E.A.; Oliver, G.; Shah, N.; Klein, A.P.; Alewine, C.; Wang, H.; Javle, M.; Shroff, R.T.; Wolff, R.A.; et al. Family history as a marker of platinum sensitivity in pancreatic adenocarcinoma. Cancer Chemother. Pharmacol. 2015, 76, 489–498. [Google Scholar] [CrossRef]
- Lowery, M.; Wong, W.; Jordan, E.J.; Lee, J.W.; Kemel, Y.; Vijai, J.; Mandelker, D.; Zehir, A.; Capanu, M.; Salo-Mullen, E.; et al. Prospective Evaluation of Germline Alterations in Patients with Exocrine Pancreatic Neoplasms. JNCI J. Natl. Cancer Inst. 2018, 110, 1067–1074. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/ PALB2 mutation. J. Clin. Oncol. 2020, 38, 1378. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Rahib, L.; Lyons, E.; De Arbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sohal, D.P.S.; et al. Outcomes in Patients with Pancreatic Adenocarcinoma with Genetic Mutations in DNA Damage Re-sponse Pathways: Results from the Know Your Tumor Program. JCO Precis. Oncol. 2019, 3, 1–10. [Google Scholar]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kanai, M.; Kou, T.; Sakuma, T.; Mochizuki, H.; Kamada, M.; Nakatsui, M.; Uza, N.; Kodama, Y.; Masui, T.; et al. Association between homologous recombination repair gene mutations and response to oxaliplatin in pancreatic cancer. Oncotarget 2018, 9, 19817–19825. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.A.; Yu, S.; Judy, R.; Symecko, H.; Nathanson, K.L.; Domchek, S.M. Retrospective Survival Analysis of Patients with Advanced Pancreatic Ductal Adenocarcinoma and Germline BRCA or PALB2 Mutations. JCO Precis. Oncol. 2018, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pokataev, I.; Fedyanin, M.; Polyanskaya, E.; Popova, A.; Agafonova, J.; Menshikova, S.; Tryakin, A.; Rumyantsev, A.; Tjulandin, S. Efficacy of platinum-based chemotherapy and prognosis of patients with pancreatic cancer with homologous recombination deficiency: Comparative analysis of published clinical studies. ESMO Open 2020, 5, e000578. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.; Mayer, E.L.; He, L.; Traina, T.A.; Carey, L.; Krag, K.J.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. TBCRC009: A multicenter phase II clinical trial of platinum monotherapy with biomarker assessment in metastatic triple-negative breast cancer. J. Clin. Oncol. 2015, 33, 1902–1909. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Penson, R.T.; Domchek, S.M.; Kaufman, B.; Shapira-Frommer, R.; Audeh, M.W.; Kaye, S.; Molife, L.R.; Gelmon, K.A.; Robertson, J.D.; et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann. Oncol. 2016, 27, 1013–1019. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncologist 2020, 26, e164–e172. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Tutt, A.N.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M. Advances in the Management of Pancreatic Adenocarcinoma. J. Natl. Compr. Cancer Netw. 2020, 18, 958–961. [Google Scholar] [CrossRef]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Scher, H.I.; Sandhu, S.; Efstathiou, E.; Lara, P.N.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; Ståhl, O.; et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men with Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Meta-static Castration-Resistant Prostate Cancer: Analysis from the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef]
- Carreira, S.; Porta, N.; Arce-Gallego, S.; Seed, G.; Llop-Guevara, A.; Bianchini, D.; Rescigno, P.; Paschalis, A.; Bertan, C.; Baker, C.; et al. Biomarkers Associating with PARP Inhibitor Benefit in Prostate Cancer in the TOPARP-B Trial. Cancer Discov. 2021, 11, 2812–2827. [Google Scholar] [CrossRef]
- Ji, W.; Weng, X.; Xu, D.; Cai, S.; Lou, H.; Ding, L. Non-small cell lung cancer cells with deficiencies in homologous recombi-nation genes are sensitive to PARP inhibitors. Biochem. Biophys. Res. Commun. 2020, 522, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Corti, G.; Durinikova, E.; Montone, M.; Reilly, N.M.; Russo, M.; Lorenzato, A.; Arcella, P.; Lazzari, L.; Rospo, G.; et al. A Subset of Colorectal Cancers with Cross-Sensitivity to Olaparib and Oxaliplatin. Clin. Cancer Res. 2019, 26, 1372–1384. [Google Scholar] [CrossRef]
- Oza, J.; Doshi, S.D.; Hao, L.; Musi, E.; Schwartz, G.K.; Ingham, M. Homologous recombination repair deficiency as a therapeutic target in sarcoma. Semin. Oncol. 2020, 47, 380–389. [Google Scholar] [CrossRef]
- Dieras, V.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Veliparib with carboplatin and paclitaxel in BRCA mutated advanced breast cancer (BROCADE3): A randomized, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1269–1282. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Schwartzberg, L.; Danso, M.A.; Miller, K.D.; Rugo, H.S.; Neubauer, M.; Robert, N.; Hellerstedt, B.; Saleh, M.; Richards, P.; et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2020, 32, 3840–3847. [Google Scholar] [CrossRef]
- Coleman, R.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Ben-Baruch, N.E.; Werner, T.L.; et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Coleman, R.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomized, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Lederman, J.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomized, placebo-controlled, phase 3 trial. Lancet. Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Chakravarty, D.; Johnson, A.; Sklar, J.; Lindeman, N.I.; Moore, K.; Ganesan, S.; Lovly, C.M.; Perlmutter, J.; Gray, S.W.; Hwang, J.; et al. Somatic Genomic Testing in Patients with Metastatic or Advanced Cancer: ASCO Provisional Clinical Opinion. J. Clin. Oncol. 2022, 40, 1231–1258. [Google Scholar] [CrossRef]
- Parker, C.; Castro, E.; Fizazi, K.; Heidenreich, A.; Ost, P.; Procopio, G.; Tombal, B.; Gillessen, S. Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1119–1134. [Google Scholar] [CrossRef]
- Horgan, D.; Ciliberto, G.; Conte, P.; Curigliano, G.; Seijo, L.; Montuenga, L.M.; Garassino, M.; Penault-Llorca, F.; Galli, F.; Ray-Coquard, I.; et al. Bringing Onco-Innovation to Europe’s Healthcare Systems: The Potential of Biomarker Testing, Real World Evidence, Tumour Agnostic Therapies to Empower Personalised Medicine. Cancers 2021, 13, 583. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic (Version 2.2022). 9 March 2022. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed on 5 June 2022).
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.; Barlesi, F.; Lolkema, M.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Curtin, N.J.; Drew, Y.; Saha, S.S. Why BRCA mutations are not tumour-agnostic biomarkers for PARP inhibitor therapy. Nat. Rev. Clin. Oncol. 2019, 16, 725–726. [Google Scholar] [CrossRef]
- Miller, R.; Leary, A.; Scott, C.; Serra, V.; Lord, C.; Bowtell, D.; Chang, D.; Garsed, D.; Jonkers, J.; Ledermann, J.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef]
- Golan, T.; O’Kane, G.M.; Denroche, R.E.; Raitses-Gurevich, M.; Grant, R.C.; Holter, S.; Wang, Y.; Zhang, A.; Jang, G.H.; Stossel, C.; et al. Genomic Features and Classification of Homologous Recombination Deficient Pancreatic Ductal Adenocar-cinoma. Gastroenterology 2021, 160, 2119–2132. [Google Scholar] [CrossRef]
- How, J.; Jazaeri, A.; Fellman, B.; Daniels, M.; Penn, S.; Solimeno, C.; Yuan, Y.; Schmeler, K.; Lanchbury, J.; Timms, K.; et al. Modification of Homologous Recombination Deficiency Score Threshold and Association with Long-Term Survival in Epithelial Ovarian Cancer. Cancers 2021, 13, 946. [Google Scholar] [CrossRef]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Borcsok, J.; Pomerantz, M.M.; Tisza, V.; Spisak, S.; Rusz, O.; Csabai, I.; Freedman, M.L.; et al. Detection of Molecular Signatures of Homologous Recombination Deficiency in Prostate Cancer with or without BRCA1/2 Mutations. Clin. Cancer Res. 2020, 26, 2673–2680. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Plummer, E.R.; Elattar, A.; Soohoo, S.; Uzir, B.; Quinn, J.E.; McCluggage, W.G.; Maxwell, P.; Aneke, H.; Curtin, N.J.; et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: Sensitivity to PARP inhibitors, platinum, and survival. Cancer Res. 2012, 72, 5675–5682. [Google Scholar] [CrossRef]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef]
- Wattenberg, M.M.; Reiss, K.A. Determinants of Homologous Recombination Deficiency in Pancreatic Cancer. Cancers 2021, 13, 4716. [Google Scholar] [CrossRef] [PubMed]
- Tobalina, L.; Armenia, J.; Irving, E.; O’Connor, M.; Forment, J. A meta-analysis of reversion mutations in BRCA genes identifies signatures of DNA end-joining repair mechanisms driving therapy resistance. Ann. Oncol. 2020, 32, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Schonhoft, J.D.; Zhao, J.L.; Jendrisak, A.; Carbone, E.A.; Barnett, E.S.; Hullings, M.A.; Gill, A.; Sutton, R.; Lee, J.; Dago, A.E.; et al. Morphology-Predicted Large-Scale Transition Number in Circulating Tumor Cells Identifies a Chromosomal Instability Biomarker Associated with Poor Outcome in Castration-Resistant Prostate Cancer. Cancer Res. 2020, 80, 4892–4903. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Type of Tumour | Author | Type of Study | N | Primary Endpoint | Platinum | Benefit | Target | Sub-Population |
|---|---|---|---|---|---|---|---|---|
| Breast Cancer | ||||||||
| Localized | Tung 2020 [45] | Randomized Phase II | 118 | pCR | Cisplatin | Platinum vs. Control: 18% vs. 26%. Risk ratio 0.70 (90% CI, 0.39–1.2). | HER2- I-III | 69% gBRCA1 30% gBRCA2 2% Both |
| Hahnen 2017 [47] | Randomized Phase II | 50 | pCR | Carboplatin | Platinum vs. Control: 65.4% vs. 66.7%. Odds ratio 0.94 (0.29–3.095), (p = 0.92). | TNBC II-III | 17% gBRCA1/2 | |
| Advanced | Isakoff 2015 [65] | Phase II | 86 | ORR | Cisplatin Carboplatin | BRCA1/2 mut vs. wild type: 54.5% vs. 19.7%, (p = 0.022). | TNBC metastatic or locally recurrent unresectable | 13% gBRCA1/2 77% wild type 10% not known |
| Pancreatic Cancer | ||||||||
| Localized | Golan 2020 [48] | Retrospective analysis | 61 | pCR | Oxaliplatin | Mutated vs. non-mutated: 44.4% vs. 10%, (p = 0.009). | Borderline resectable | 23% gBRCA2 77% gBRCA wild type |
| Metastatic | Okano 2020 [49] | Phase II | 43 | OS | Oxaliplatin | 1-year survival 27.9% (90% CI 17–41.3). Primary endpoint not met (30%). | Metastatic PDAC | Family history (ovarian, prostate, pancreatic, breast)
|
| Wattenberg 2020 [50] | Retrospective analysis | 26 | PFS | Oxaliplatin Cisplatin | Mutated vs. non-mutated: 10.1 vs. 6.9 months, (p = 0.0068) | Locally advanced or metastatic | 33% Mutated:
| |
| Prostate Cancer | ||||||||
| Castration-resistant prostate cancer | Schmid 2020 [51] | Retrospective analysis | 508 | Platinum Antitumor activity (decrease PSA 50% and/or radiological response) | Carboplatin Cisplatin Oxaliplatin | Mutated (cohort 1) vs. non-mutated (cohort 2) decrease PSA: 47.1% vs. 36.1%, (p = 0.20). | Advanced |
|
| Mota 2020 [52] | Retrospective analysis | 109 | Platinum efficacy in DDR-mutant | Carboplatin Cisplatin | 67% BRCA2 achieved a PSA50 response (adjusted Odds Ratio 9.5; 95% CI 1.5–82.9) compared to DDRwt (13%), (p = 0.022). | Metastatic |
| |
| Type of Tumor | Author | Principal Endpoint | Treatment | Benefit | OS Benefit | Target | Sub-Population |
|---|---|---|---|---|---|---|---|
| Breast Cancer | |||||||
| Localized disease | Tutt et al., 2021 [80] | DFS | Local treatment and neoadjuvant or adjuvant chemotherapy. Olaparib vs. placebo. | Yes | NS | gBRCA1/2 | 71.3% BRCA1 28.3% BRCA2 |
| Pre-treated M1 or unresectable | Diéras 2020 [93] | PFS | Carbo, pacli ± veliparib | Yes | NS | gBRCA1/2 | - |
| Litton 2018 [78] | PFS | Chemo 1 vs. Talazoparib | Yes | NS | gBRCA1/2 | - | |
| Robson 2017 [79] | PFS | Chemo 1 vs. olaparib | Yes | NS | gBRCA1/2 | - | |
| O’Shaughnessy 2014 [94] | PFS and OS | Carbo, gem ± iniparib | Yes | Yes | Triple negative | ||
| Ovarian Cancer | |||||||
| 1st line maintenance | Coleman 2019 [95] | PFS | Carbo, pacli ± veliparib | Yes | NR | Platinum sensitive | 30% BRCA, 60% HRD |
| Gonzalez-Martin 2019 [76] | PFS | Niraparib vs. placebo | Yes | NS 2 | Platinum sensitive | 30% BRCA 51% HRD | |
| Ray-Coquard 2019 [76] | PFS | Olaparib + Bevacizumab | Yes | NR | Platinum sensitive | 30% BRCA 50% HRD | |
| Moore 2018 [74] | PFS | Olaparib vs. placebo | Yes | NS 2 | BRCA1/23 | ||
| Platinum sensitive recurrence | Coleman 2017 [96] | PFS | Rucaparib vs. placebo | Yes | NS 2 | Platinum sensitive | 35% BRCA 60% HRD |
| Pujade-Lauraine 2017 [97] | PFS | Olaparib vs. placebo | Yes | NS | gBRCA1/2 | ||
| Mirza 2016 [72] | PFS | Niraparib vs. placebo | Yes | NS | Platinum sensitive | BRCA and non-BRCA cohorts | |
| Pancreatic Cancer | |||||||
| 1st line maintenance | Golan 2019 [61] | PFS | Olaparib vs. placebo | Yes | NS 2 | gBRCA1/2 + platinum sensitive | |
| Prostate Cancer | |||||||
| Pre-treated M1 CRPC | De Bono 2020 [83] | PFS in cohort A | Olaparib vs. AA/enza | Yes | NS | Somatic HRD by NGS 15 genes multi-panel | Cohort A: BRCA + ATM Cohort B: non-BRCA/ATM |
| Breast and/or Ovarian Cancer | Exocrine Pancreatic Cancer | Prostate Cancer | |
|---|---|---|---|
| Hereditary testing criteria | All patients diagnosed with epithelial ovarian cancer (including fallopian or peritoneal cancer). Any blood relative with a known pathogenic/likely pathogenic variant. Personal history of breast cancer with specific features:
| All individuals diagnosed. First-degree relatives of individuals diagnosed * | Metastatic prostate cancer Intraductal/cribriform histology High or very high-risk group Family history:
|
| Genetic testing process | |||
| -Familial pathogenic/likely pathogenic variant known | Testing for specific familial pathogenic/likely pathogenic variant | Testing for specific familial variant.
| Consider NGS panel testing. |
| -No known familial pathogenic/likely pathogenic variant | Comprehensive testing with multigene panel | Comprehensive testing with multigene panel. | In the abscense of family history or clinical features may be of low yield. |
| Germline recommendations | BRCA1, BRCA2, ATM, BARD1, BRIP1, CDH1, CDKN2A, CHEK2, NBN, NF1, PALB2, PTEN, RAD51C, RAD51D, STK11, TP53. Lynch syndrome genes (MLH1, MSH2, MSH6, PM2). | BRCA1, BRCA2, ATM, CDKN2A, Lynch syndrome genes (MLH1, MSH2, MSH6, EPCAM), PALB2, STK11 and TP53. | BRCA1, BRCA2, ATM, PABL2, CHECK2. Lynch syndrome genes (MLH1, MSH2, MSH6, PM2).
|
| Somatic testing ASCO recommendations | BRCA 1/2 NTRK1, NTRK2, NTRK3 fusions MSI-H, TMB-H | ||
| Breast cancer: ERBB2 amplification. Oncogenic mutations in PIK3CA in HR+ HER2−. Ovarian cancer: GIS-positive or HRD-positive. | MSI-H, ATM, BARD1, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, RAD51B, RAD51C, RAD51D, RAD54L. | ||
| ESMO Scale for clinical actionability of molecular targets | Metastatic breast cancer: BRCA1/2 (germline/somatic) | Advanced pancreatic cancer: BRCA1/2 germline/somatic mutations, MSI-H | Advanced prostate cancer: BRCA1/2 somatic mutations/deletions, MSI-H, ATM mutations/deletions |
| NGS recommendations | Tumour multigene NGS can be used in ovarian cancer to determine somatic BRCA1/2. In breast cancer, no current indication for tumour multigene NGS. | No current indication for tumour multigene NGS | Multigene tumour NGS to assess level I alterations. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacheco-Barcia, V.; Muñoz, A.; Castro, E.; Ballesteros, A.I.; Marquina, G.; González-Díaz, I.; Colomer, R.; Romero-Laorden, N. The Homologous Recombination Deficiency Scar in Advanced Cancer: Agnostic Targeting of Damaged DNA Repair. Cancers 2022, 14, 2950. https://doi.org/10.3390/cancers14122950
Pacheco-Barcia V, Muñoz A, Castro E, Ballesteros AI, Marquina G, González-Díaz I, Colomer R, Romero-Laorden N. The Homologous Recombination Deficiency Scar in Advanced Cancer: Agnostic Targeting of Damaged DNA Repair. Cancers. 2022; 14(12):2950. https://doi.org/10.3390/cancers14122950
Chicago/Turabian StylePacheco-Barcia, Vilma, Andrés Muñoz, Elena Castro, Ana Isabel Ballesteros, Gloria Marquina, Iván González-Díaz, Ramon Colomer, and Nuria Romero-Laorden. 2022. "The Homologous Recombination Deficiency Scar in Advanced Cancer: Agnostic Targeting of Damaged DNA Repair" Cancers 14, no. 12: 2950. https://doi.org/10.3390/cancers14122950
APA StylePacheco-Barcia, V., Muñoz, A., Castro, E., Ballesteros, A. I., Marquina, G., González-Díaz, I., Colomer, R., & Romero-Laorden, N. (2022). The Homologous Recombination Deficiency Scar in Advanced Cancer: Agnostic Targeting of Damaged DNA Repair. Cancers, 14(12), 2950. https://doi.org/10.3390/cancers14122950