
Generation of the Chondroprotective Proteomes by Activating PI3K and TNFα Signaling

 ,
,  , and
, and 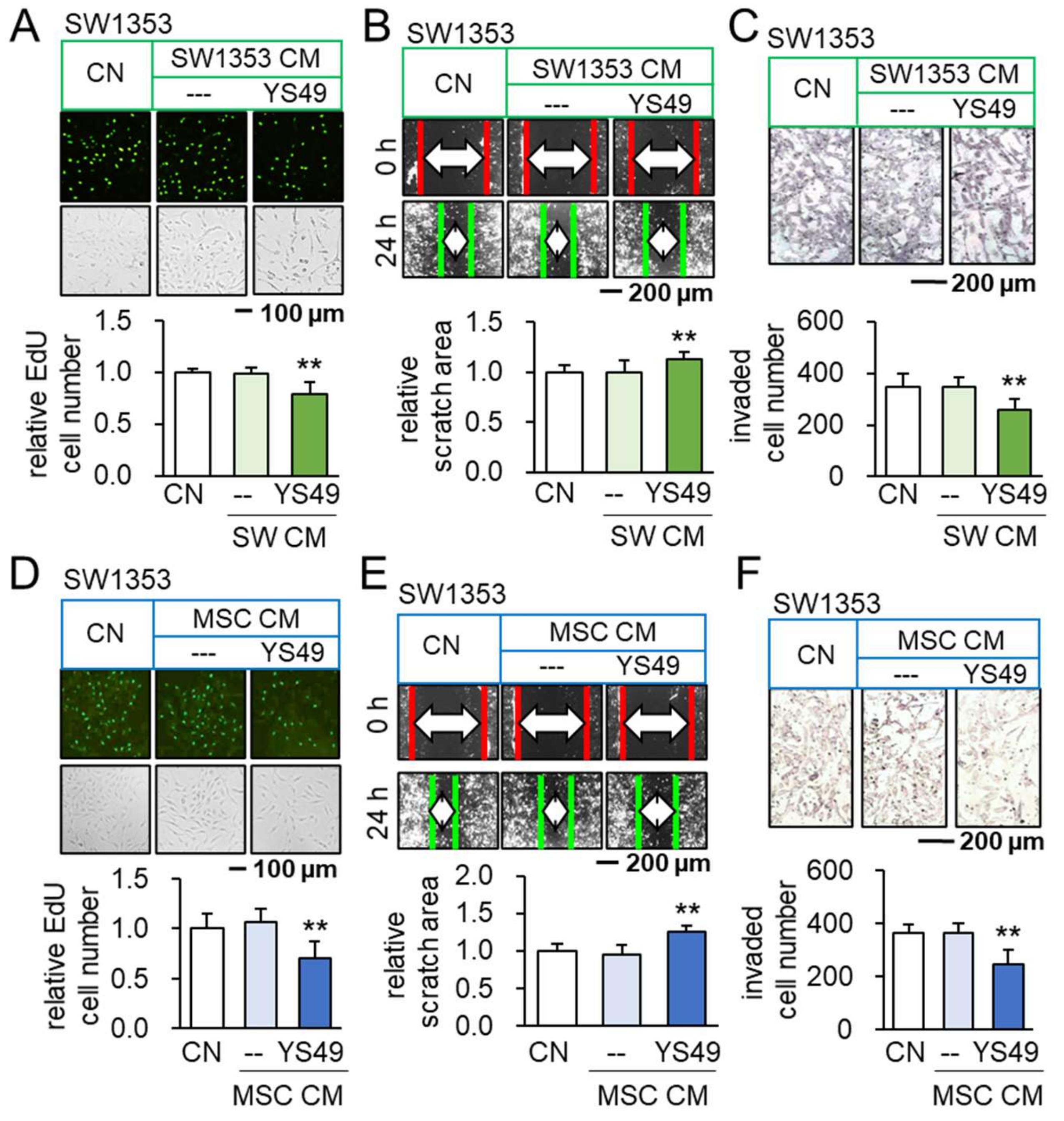{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Agents
2.3. Preparation of Conditioned Medium (CM) and ELISA Assay
2.4. MTT and EdU Assays
2.5. Transwell Invasion and Scratch Motility Assays
2.6. Western Blot Analysis and Mass Spectrometry
2.7. Immunoprecipitation
2.8. Plasmid Transfection and RNA Interference
2.9. Animal Models
2.10. µ. CT Imaging and Histology
2.11. Statistical Analysis
3. Results
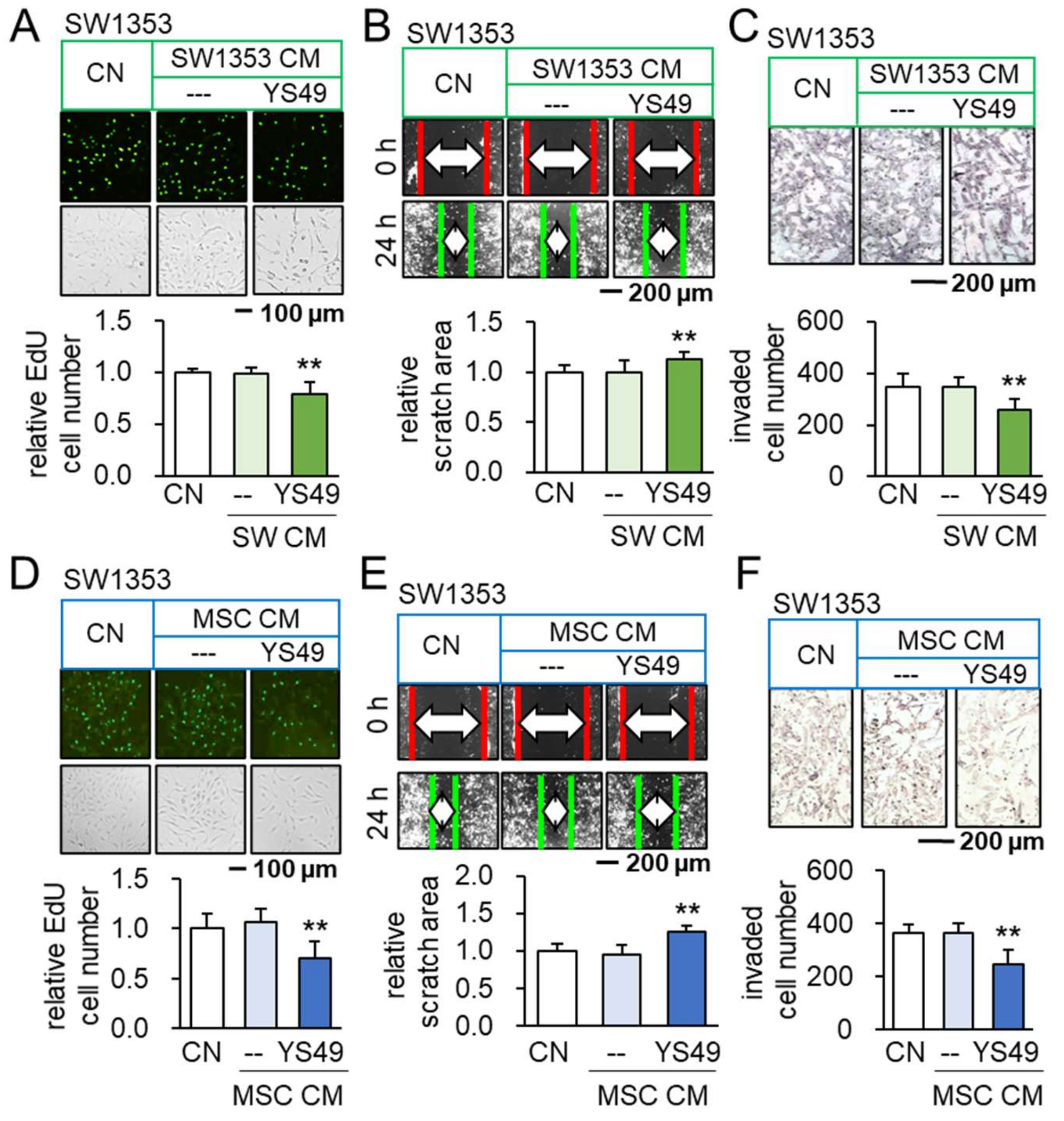
3.1. Anti-Tumorigenic Effects of YS49-Treated SW1353/MSC-Derived CM
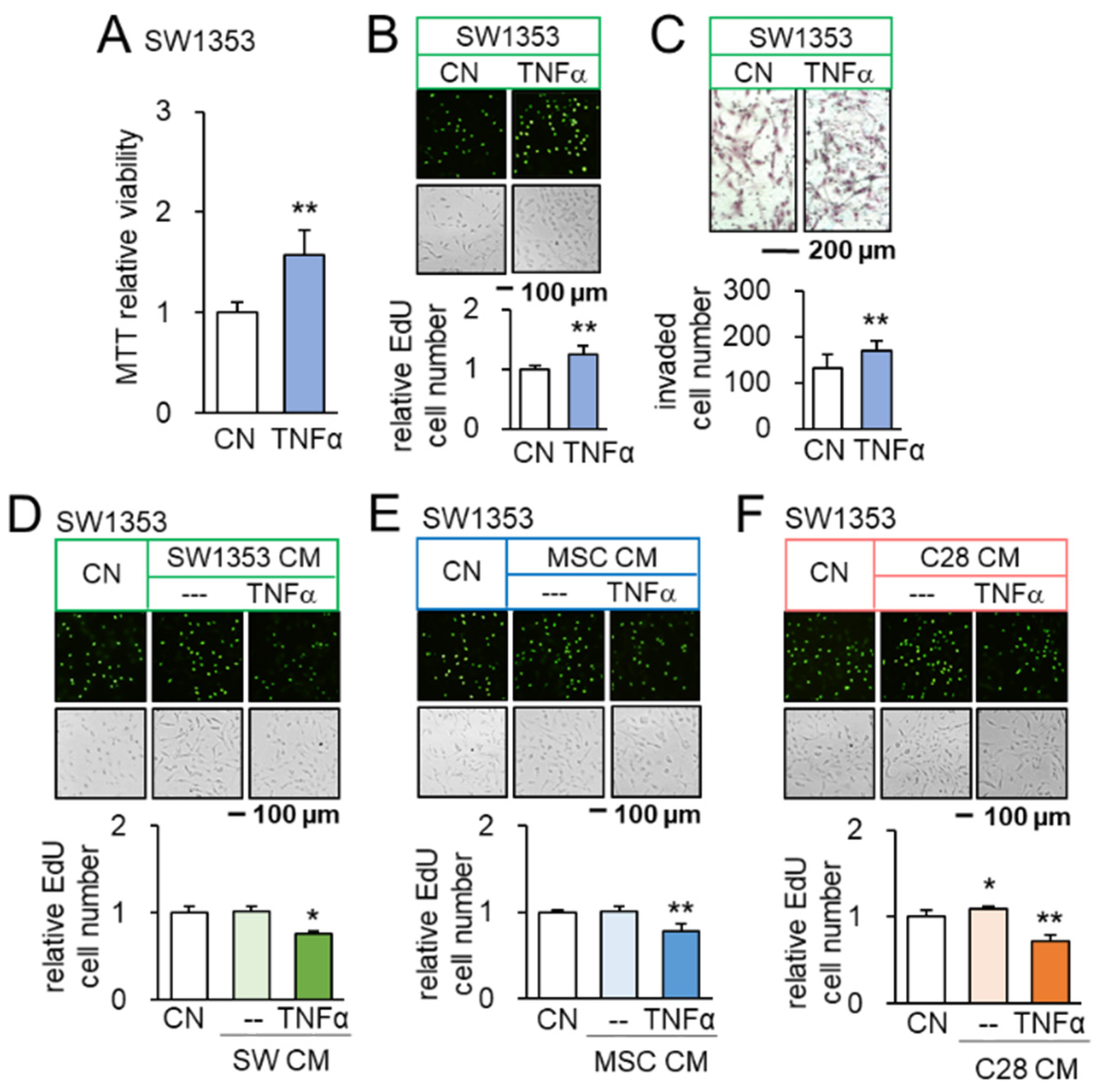
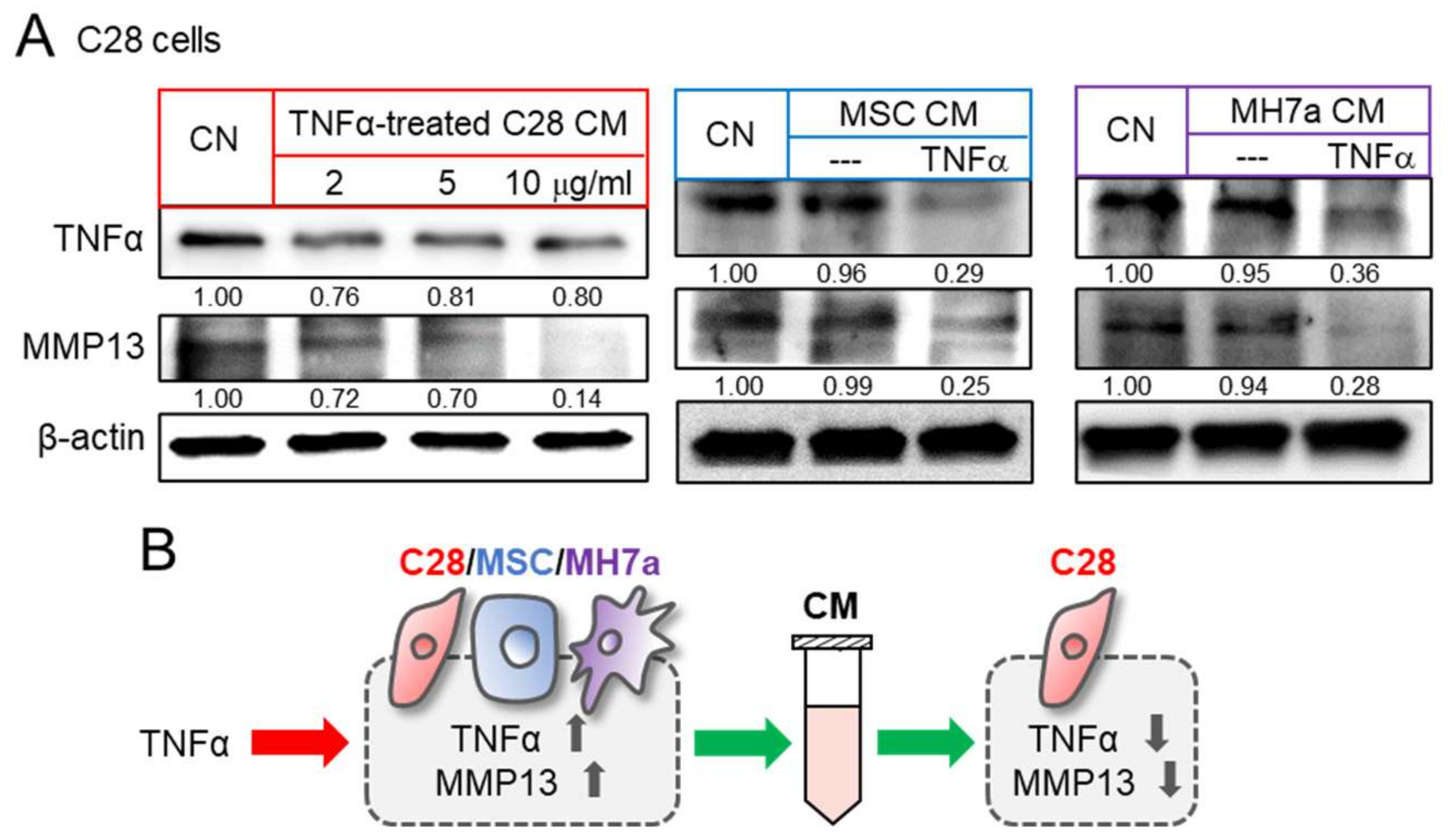
3.2. Promotion of Tumorigenic Responses by TNFα
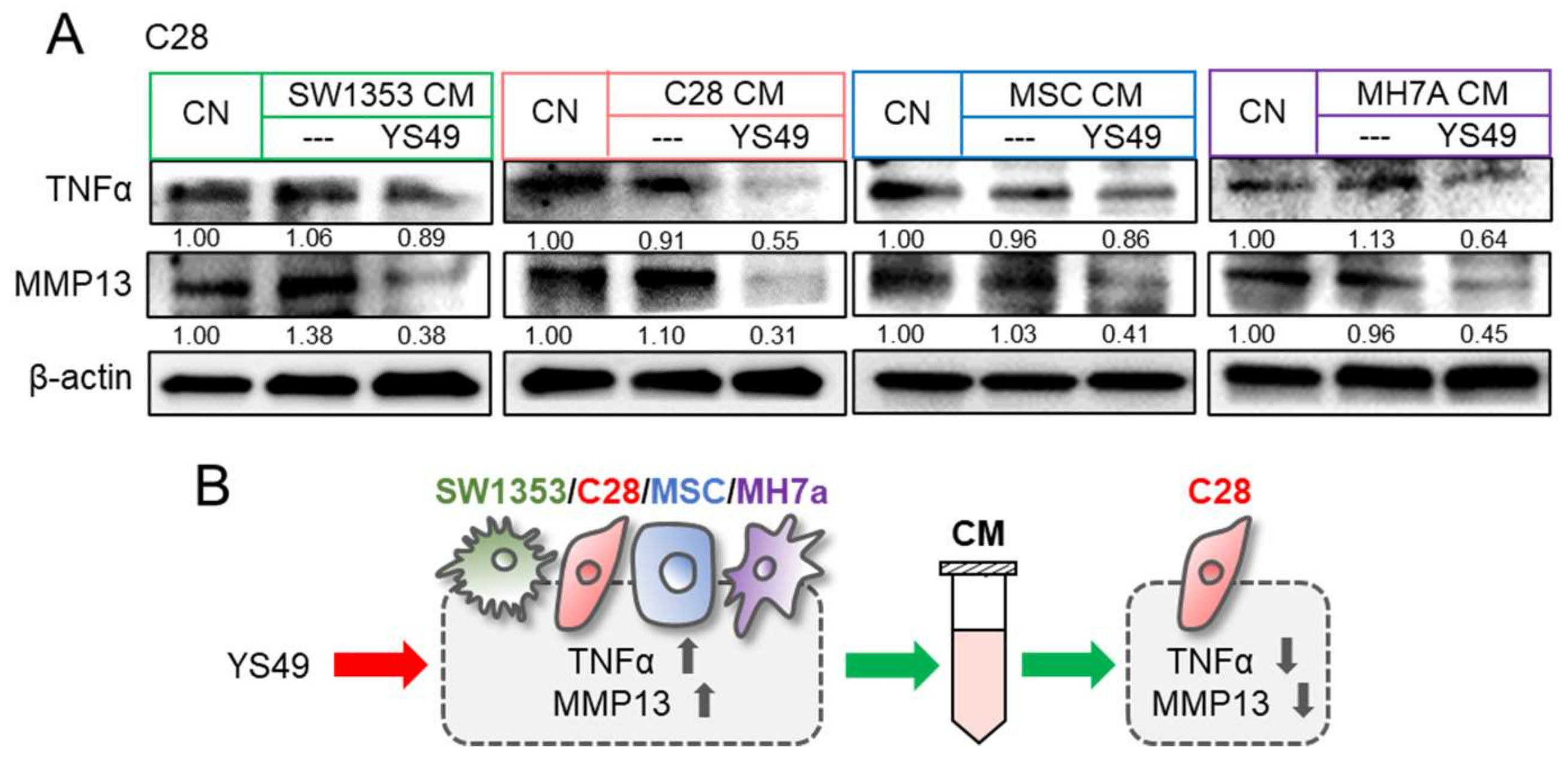
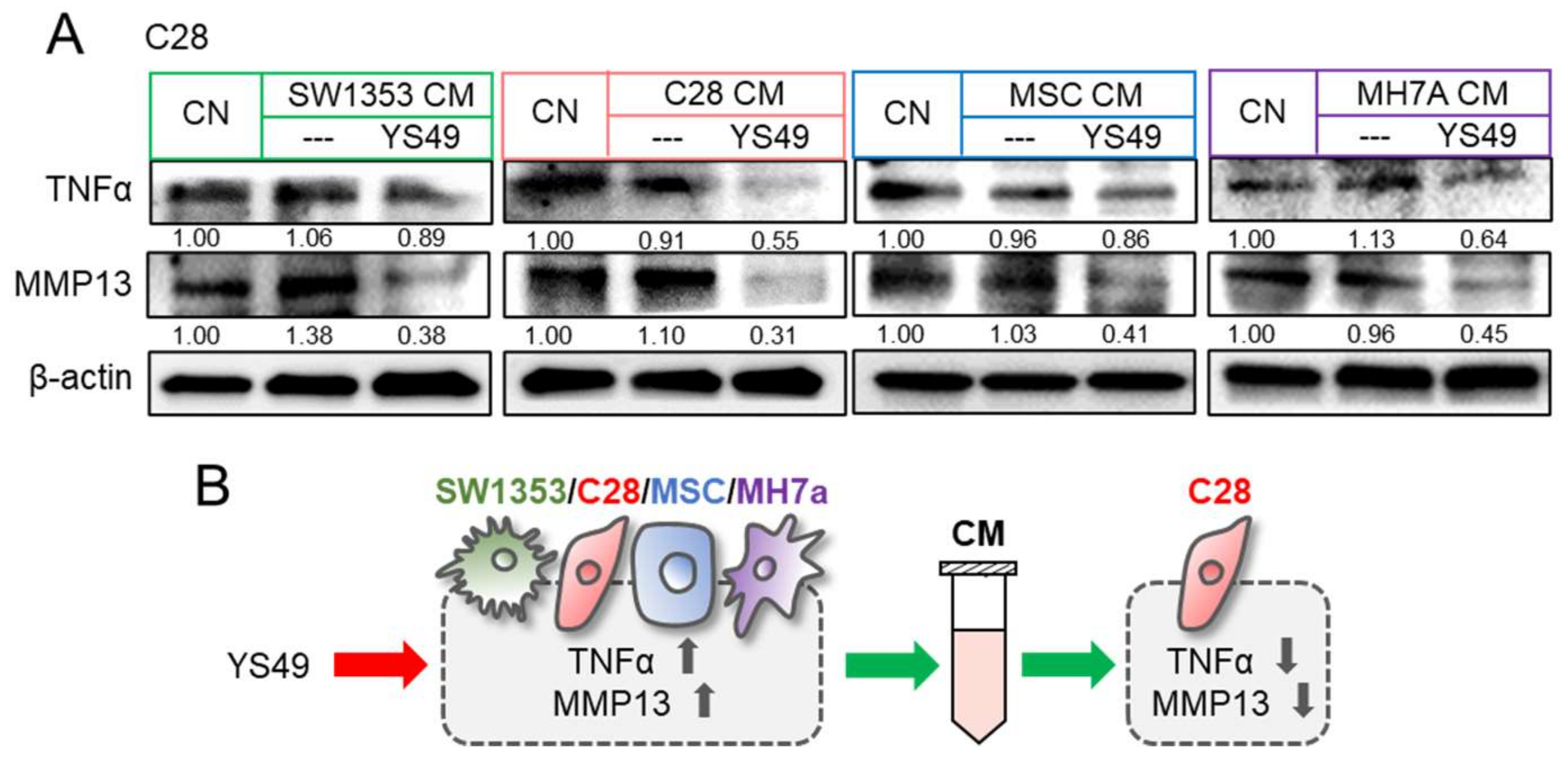
3.3. Generation of Anti-Inflammatory CM
3.4. Context-Dependent Effects by the Activation of PI3K/Akt Signaling
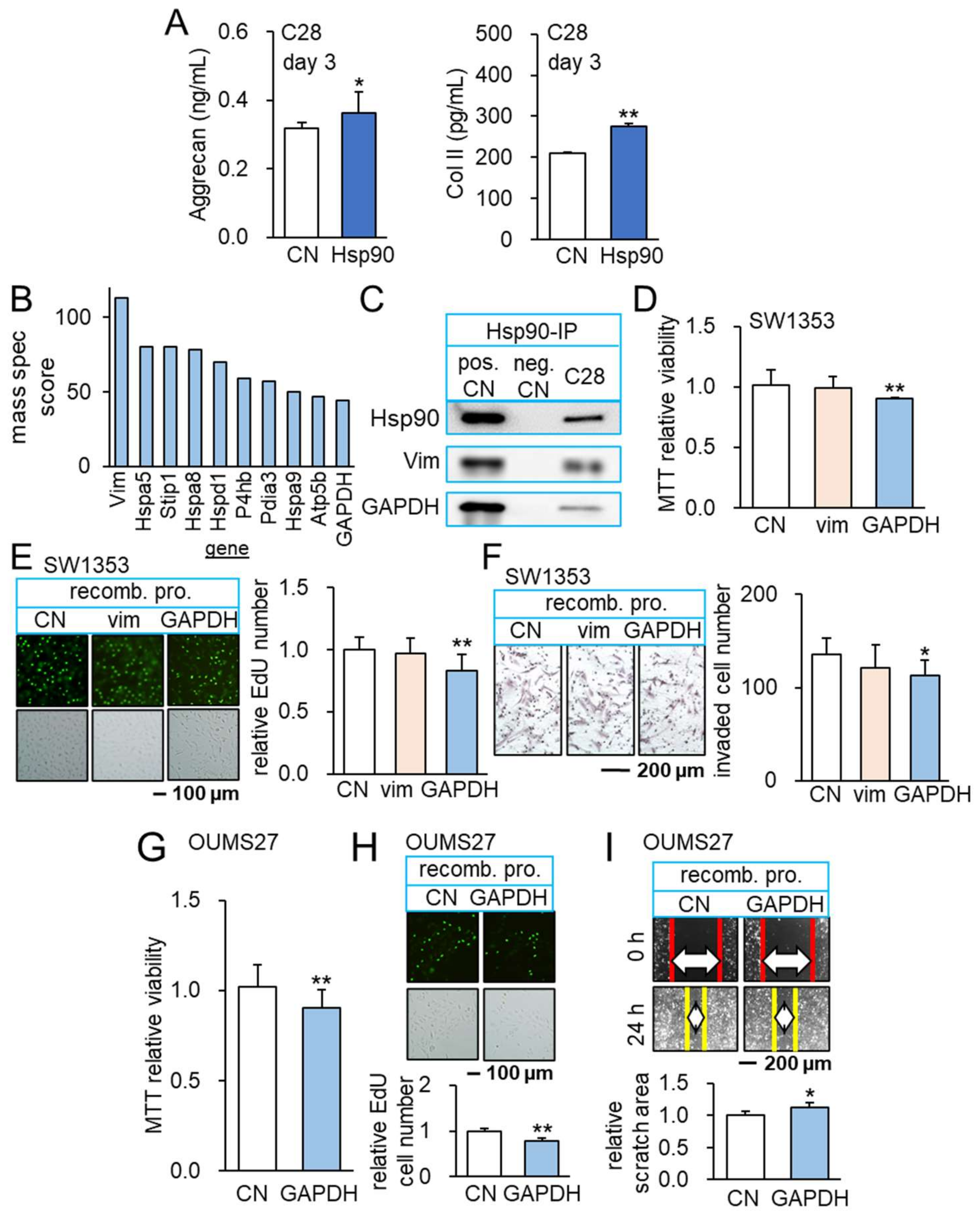
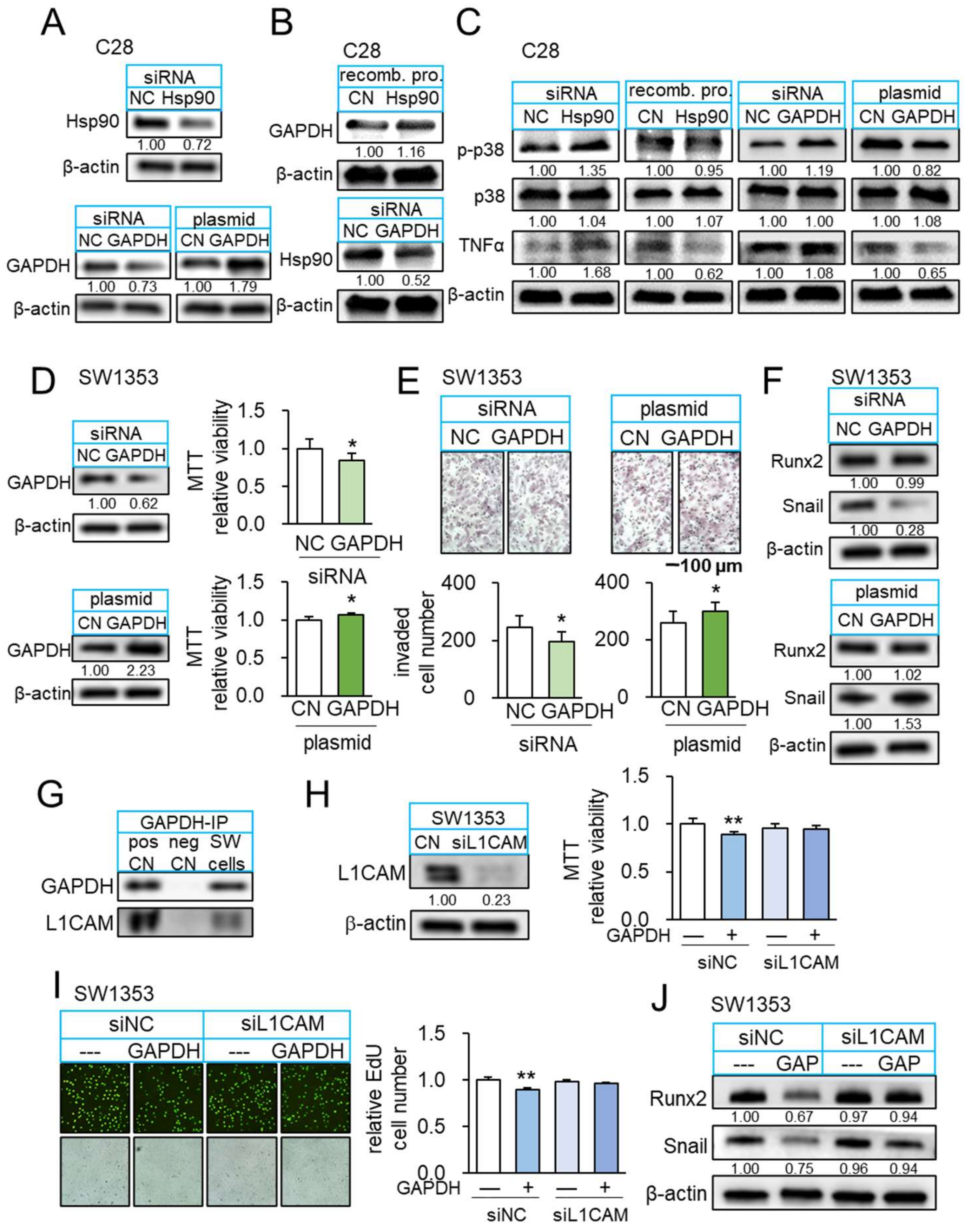
3.5. Beneficial Role of Extracellular Hsp90ab1
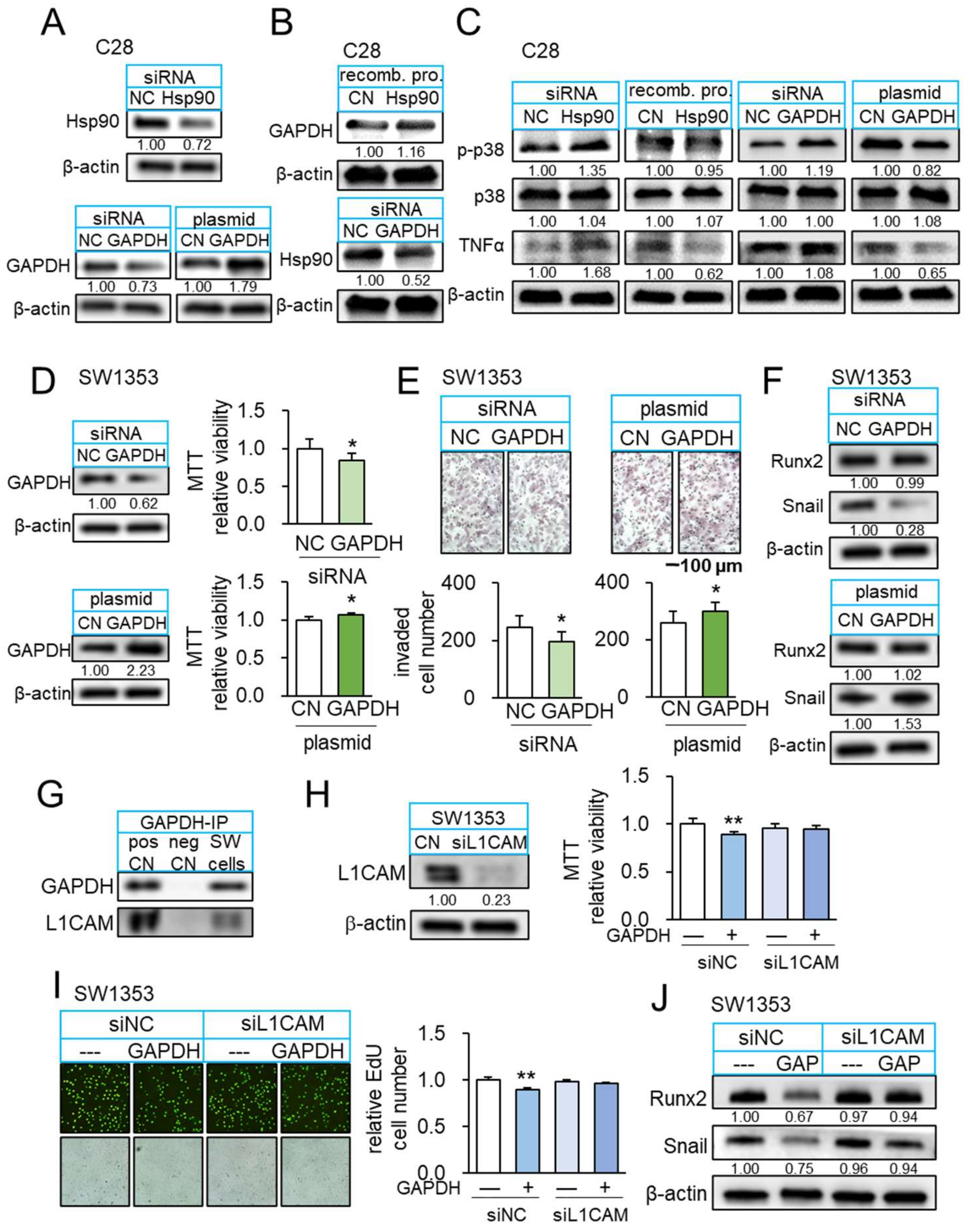
3.6. Interaction of Hsp90ab1 with Extracellular GAPDH
3.7. GAPDH as an Anti-Inflammatory Protein
3.8. GAPDH as an Anti-Tumor Protein and Its Interaction with L1CAM
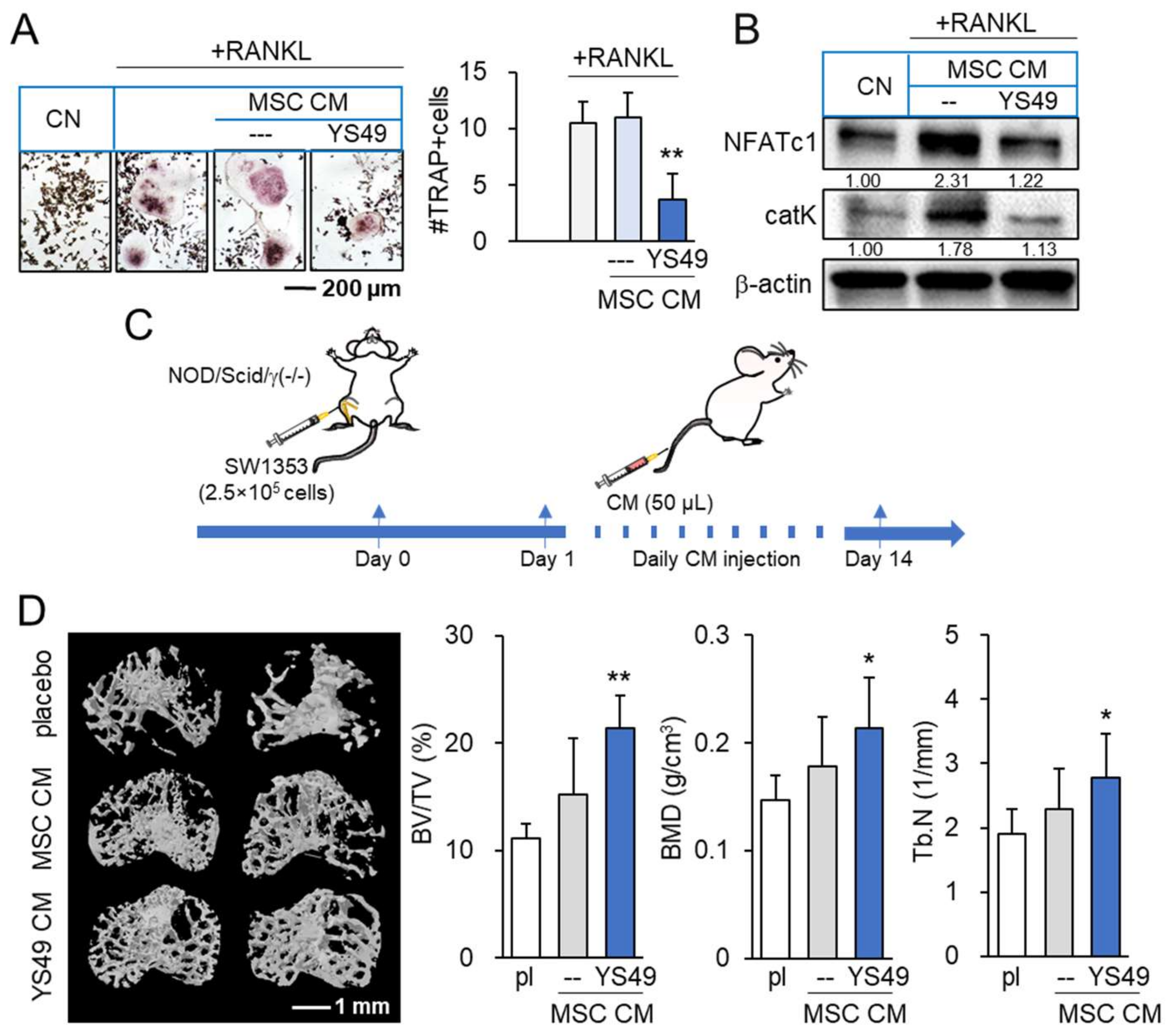
3.9. Suppression of Osteoclast Development and Tumor-Driven Bone Loss by YS MSC CM
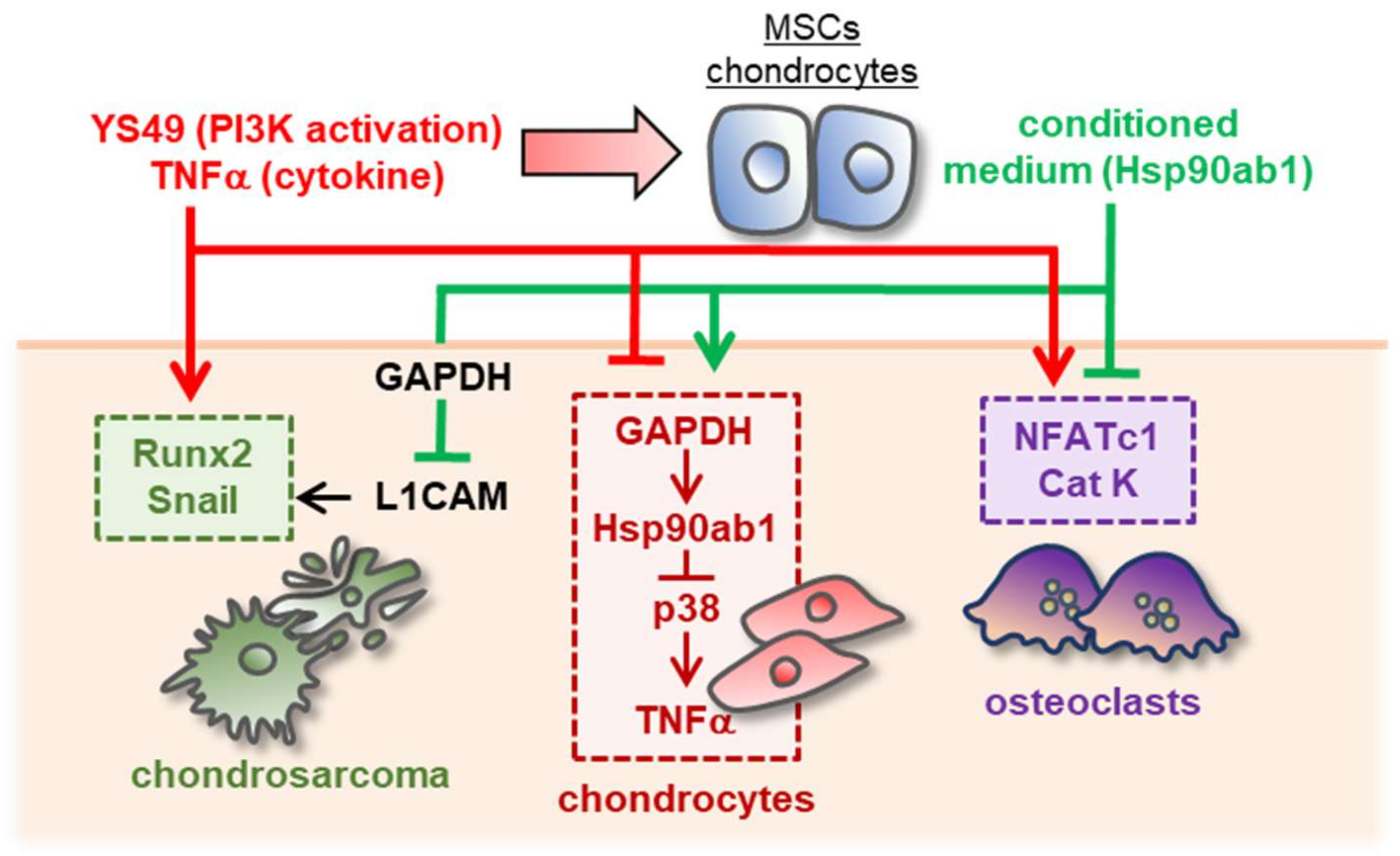
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Qiao, D.; Chen, L.; Xu, M.; Chen, S.; Huang, L.; Wang, F.; Chen, Z.; Cai, J.; Fu, L. Chemotherapeutic drugs stimulate the release and recycling of extracellular vesicles to assist cancer cells in developing an urgent chemoresistance. Mol. Cancer 2019, 18, 182. [Google Scholar] [CrossRef]
- Parker, T.M.; Henriques, V.; Beltran, A.; Nakshatri, H.; Gogna, R. Cell competition and tumor heterogeneity. Semin. Cancer Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, J. Cancer and arthritis share underlying processes. J. Natl. Cancer Inst. 1998, 90, 802–803. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, Z.; Bai, L. Cell Interplay in Osteoarthritis. Front. Cell Dev. Biol. 2021, 9, 720477. [Google Scholar] [CrossRef] [PubMed]
- Murphey, M.D.; Walker, E.A.; Wilson, A.J.; Kransdorf, M.J.; Temple, H.T.; Gannon, F.H. From the archives of the AFIP: Imaging of primary chondrosarcoma: Radiologic-pathologic correlation. Radiographics 2003, 23, 1245–1278. [Google Scholar] [CrossRef]
- Bullock, J.; Rizvi, S.A.A.; Saleh, A.M.; Ahmed, S.S.; Do, D.P.; Ansari, R.A.; Ahmed, J. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med. Princ. Pract. 2018, 27, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.; Hogendoorn, P.C.; Dijkstra, S.D.; van Rijswijk, C.S.; Krol, A.D.; Taminiau, A.H.; Bovée, J.V. The clinical approach towards chondrosarcoma. Oncologist 2008, 13, 320–329. [Google Scholar] [CrossRef]
- Köhler, B.M.; Günther, J.; Kaudewitz, D.; Lorenz, H.M. Current Therapeutic Options in the Treatment of Rheumatoid Arthritis. J. Clin. Med. 2019, 8, 938. [Google Scholar] [CrossRef]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef]
- Bhatia, D.; Bejarano, T.; Novo, M. Current interventions in the management of knee osteoarthritis. J. Pharm. Bioallied Sci. 2013, 5, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Nagata, R.; Nakamura, M.; Sanaki, Y.; Igaki, T. Cell Competition Is Driven by Autophagy. Dev. Cell 2019, 51, 99–112. [Google Scholar] [CrossRef]
- Giraldez, A.J.; Cohen, S.M. Wingless and Notch signaling provide cell survival cues and control cell proliferation during wing development. Development 2003, 130, 6533–6543. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.A.; Sanders, A.L. Wingless promotes cell survival but constrains growth during Drosophila wing development. Nat. Cell Biol. 2003, 5, 827–833. [Google Scholar] [CrossRef] [PubMed]
- de la Cova, C.; Abril, M.; Bellosta, P.; Gallant, P.; Johnston, L.A. Drosophila myc regulates organ size by inducing cell competition. Cell 2004, 117, 107–116. [Google Scholar] [CrossRef]
- Senoo-Matsuda, N.; Johnston, L.A. Soluble factors mediate competitive and cooperative interactions between cells expressing different levels of Drosophila Myc. Proc. Natl. Acad. Sci. USA 2007, 104, 18543–18548. [Google Scholar] [CrossRef]
- Liu, S.; Sun, X.; Li, K.; Zha, R.; Feng, Y.; Sano, T.; Dong, C.; Liu, Y.; Aryal, U.K.; Sudo, A.; et al. Generation of the tumor-suppressive secretome from tumor cells. Theranostics 2021, 11, 8517–8534. [Google Scholar] [CrossRef]
- Liu, S.; Wu, D.; Sun, X.; Fan, Y.; Zha, R.; Jalali, A.; Feng, Y.; Li, K.; Sano, T.; Vike, N.; et al. Overexpression of Lrp5 enhanced the anti-breast cancer effects of osteocytes in bone. Bone Res. 2021, 9, 32. [Google Scholar] [CrossRef]
- Sun, X.; Li, K.; Zha, R.; Liu, S.; Fan, Y.; Wu, D.; Hase, M.; Aryal, U.K.; Lin, C.C.; Li, B.Y.; et al. Preventing tumor progression to the bone by induced tumor-suppressing MSCs. Theranostics 2021, 11, 5143–5159. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef]
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. Biochim. Biophys. Acta 2015, 1851, 882–897. [Google Scholar] [CrossRef]
- Sano, T.; Sun, X.; Feng, Y.; Liu, S.; Hase, M.; Fan, Y.; Zha, R.; Wu, D.; Aryal, U.K.; Li, B.Y.; et al. Inhibition of the Growth of Breast Cancer-Associated Brain Tumors by the Osteocyte-Derived Conditioned Medium. Cancers 2021, 13, 1061. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.D.; Harmer, D.W.; Coleman, R.A.; Clark, B.J. GAPDH as a housekeeping gene: Analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol. Genom. 2005, 21, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.C.; Xie, Y.M.; Ran, L.Q.; Cao, H.H.; Sun, C.; Wu, J.Y.; Wu, Z.Y.; Liao, L.D.; Zhao, W.J.; Fang, W.K.; et al. L1CAM drives oncogenicity in esophageal squamous cell carcinoma by stimulation of ezrin transcription. J. Mol. Med. 2017, 95, 1355–1368. [Google Scholar] [CrossRef] [PubMed]
- Aslakson, C.J.; Miller, F.R. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992, 52, 1399–1405. [Google Scholar] [PubMed]
- Claassen, H.; Schicht, M.; Brandt, J.; Reuse, K.; Schädlich, R.; Goldring, M.B.; Guddat, S.S.; Thate, A.; Paulsen, F. C-28/I2 and T/C-28a2 chondrocytes as well as human primary articular chondrocytes express sex hormone and insulin receptors--Useful cells in study of cartilage metabolism. Ann. Anat. 2011, 193, 23–29. [Google Scholar] [CrossRef]
- Gebauer, M.; Saas, J.; Sohler, F.; Haag, J.; Söder, S.; Pieper, M.; Bartnik, E.; Beninga, J.; Zimmer, R.; Aigner, T. Comparison of the chondrosarcoma cell line SW1353 with primary human adult articular chondrocytes with regard to their gene expression profile and reactivity to IL-1beta. Osteoarthr. Cartil. 2005, 13, 697–708. [Google Scholar] [CrossRef]
- Kunisada, T.; Miyazaki, M.; Mihara, K.; Gao, C.; Kawai, A.; Inoue, H.; Namba, M. A new human chondrosarcoma cell line (OUMS-27) that maintains chondrocytic differentiation. Int. J. Cancer 1998, 77, 854–859. [Google Scholar] [CrossRef]
- Ralph, P.; Nakoinz, I. Antibody-dependent killing of erythrocyte and tumor targets by macrophage-related cell lines: Enhancement by PPD and LPS. J. Immunol. 1977, 119, 950–954. [Google Scholar]
- Miyazawa, K.; Mori, A.; Okudaira, H. Establishment and characterization of a novel human rheumatoid fibroblast-like synoviocyte line, MH7A, immortalized with SV40 T antigen. J. Biochem. 1998, 124, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Fan, Y.; Chen, A.; Jalali, A.; Minami, K.; Ogawa, K.; Nakshatri, H.; Li, B.Y.; Yokota, H. Osteocyte-Driven Downregulation of Snail Restrains Effects of Drd2 Inhibitors on Mammary Tumor Cells. Cancer Res. 2018, 78, 3865–3876. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Minami, K.; Chen, A.; Wan, Q.; Yin, Y.; Gan, L.; Xu, A.; Matsuura, N.; Koizumi, M.; et al. Inhibiting checkpoint kinase 1 protects bone from bone resorption by mammary tumor in a mouse model. Oncotarget 2018, 9, 9364–9378. [Google Scholar] [CrossRef]
- Takigawa, S.; Frondorf, B.; Liu, S.; Liu, Y.; Li, B.; Sudo, A.; Wallace, J.M.; Yokota, H.; Hamamura, K. Salubrinal improves mechanical properties of the femur in osteogenesis imperfecta mice. J. Pharmacol. Sci. 2016, 132, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.E.; Hedrick, V.; Paschoal Sobreira, T.J.; Dykhuizen, E.C.; Aryal, U.K. Analysis of Human Nuclear Protein Complexes by Quantitative Mass Spectrometry Profiling. Proteomics 2018, 18, e1700427. [Google Scholar] [CrossRef] [PubMed]
- Opoku-Temeng, C.; Onyedibe, K.I.; Aryal, U.K.; Sintim, H.O. Proteomic analysis of bacterial response to a 4-hydroxybenzylidene indolinone compound, which re-sensitizes bacteria to traditional antibiotics. J. Proteom. 2019, 202, 103368. [Google Scholar] [CrossRef]
- Makhina, T.; Loers, G.; Schulze, C.; Ueberle, B.; Schachner, M.; Kleene, R. Extracellular GAPDH binds to L1 and enhances neurite outgrowth. Mol. Cell Neurosci. 2009, 41, 206–218. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Liu, S.-Z.; Sun, X.; Li, K.-X.; Lin, C.-C.; Na, S.; Li, B.-Y.; Yokota, H. Tumor Cell Secretomes in Response to Anti- and Pro-Tumorigenic Agents. Onco 2021, 1, 101–113. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Guzhova, I.V.; Margulis, B.A. Glyceraldehyde-3-phosphate Dehydrogenase is a Multifaceted Therapeutic Target. Pharmaceutics 2020, 12, 416. [Google Scholar] [CrossRef]
- Colell, A.; Green, D.R.; Ricci, J.E. Novel roles for GAPDH in cell death and carcinogenesis. Cell Death Differ. 2009, 16, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, Y.; Goto, S.; Nakano, T.; Tseng, H.P.; Yang, S.M.; Kawamoto, S.; Ono, K.; Chen, C.L. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) prevents lipopolysaccharide (LPS)-induced, sepsis-related severe acute lung injury in mice. Sci. Rep. 2014, 4, 5204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Zhang, F.; Hong, C.Q.; Giuliano, A.E.; Cui, X.J.; Zhou, G.J.; Zhang, G.J.; Cui, Y.K. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol. Med. 2015, 12, 10–22. [Google Scholar] [CrossRef]
- Jacquin, M.A.; Chiche, J.; Zunino, B.; Bénéteau, M.; Meynet, O.; Pradelli, L.A.; Marchetti, S.; Cornille, A.; Carles, M.; Ricci, J.E. GAPDH binds to active Akt, leading to Bcl-xL increase and escape from caspase-independent cell death. Cell Death Differ. 2013, 20, 1043–1054. [Google Scholar] [CrossRef]
- Chiche, J.; Pommier, S.; Beneteau, M.; Mondragón, L.; Meynet, O.; Zunino, B.; Mouchotte, A.; Verhoeyen, E.; Guyot, M.; Pagès, G.; et al. GAPDH enhances the aggressiveness and the vascularization of non-Hodgkin’s B lymphomas via NF-κB-dependent induction of HIF-1α. Leukemia 2015, 29, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, C.; Pinto, A.R.; Li, H.; Li, L.; Wang, L.; Simpson, R.; Liu, J.P. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) induces cancer cell senescence by interacting with telomerase RNA component. Proc. Natl. Acad. Sci. USA 2012, 109, 13308–13313. [Google Scholar] [CrossRef]
- Boyle, D.L.; Jones, T.L.; Hammaker, D.; Svensson, C.I.; Rosengren, S.; Albani, S.; Sorkin, L.; Firestein, G.S. Regulation of peripheral inflammation by spinal p38 MAP kinase in rats. PLoS Med. 2006, 3, e338. [Google Scholar] [CrossRef] [PubMed]
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Li, K.-X.; Figueiredo, M.L.; Lin, C.-C.; Li, B.-Y.; Yokota, H. Generation of the Chondroprotective Proteomes by Activating PI3K and TNFα Signaling. Cancers 2022, 14, 3039. https://doi.org/10.3390/cancers14133039
Sun X, Li K-X, Figueiredo ML, Lin C-C, Li B-Y, Yokota H. Generation of the Chondroprotective Proteomes by Activating PI3K and TNFα Signaling. Cancers. 2022; 14(13):3039. https://doi.org/10.3390/cancers14133039
Chicago/Turabian StyleSun, Xun, Ke-Xin Li, Marxa L. Figueiredo, Chien-Chi Lin, Bai-Yan Li, and Hiroki Yokota. 2022. "Generation of the Chondroprotective Proteomes by Activating PI3K and TNFα Signaling" Cancers 14, no. 13: 3039. https://doi.org/10.3390/cancers14133039
APA StyleSun, X., Li, K.-X., Figueiredo, M. L., Lin, C.-C., Li, B.-Y., & Yokota, H. (2022). Generation of the Chondroprotective Proteomes by Activating PI3K and TNFα Signaling. Cancers, 14(13), 3039. https://doi.org/10.3390/cancers14133039