
Breast Cancer Genomics: Primary and Most Common Metastases

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
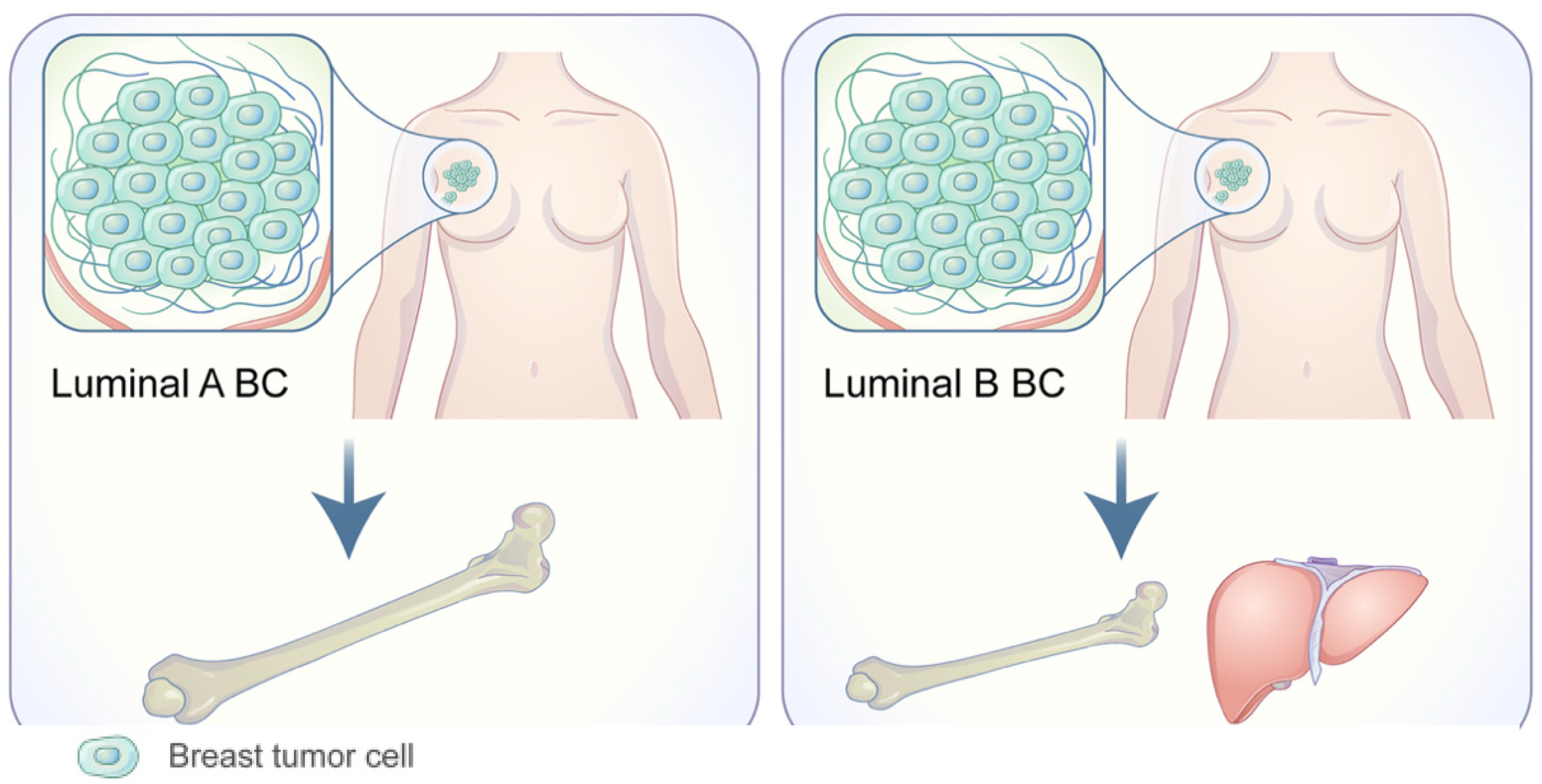
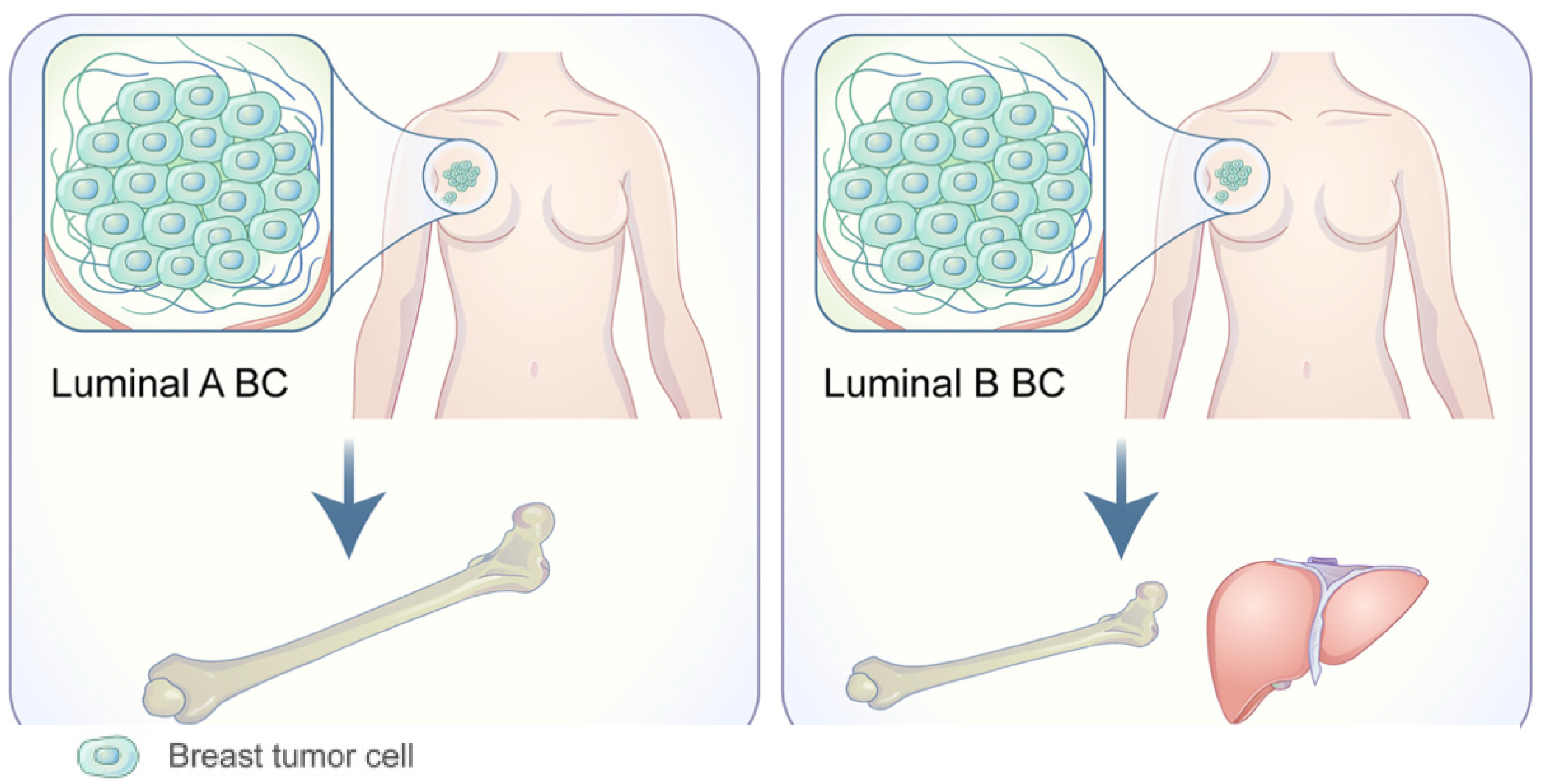
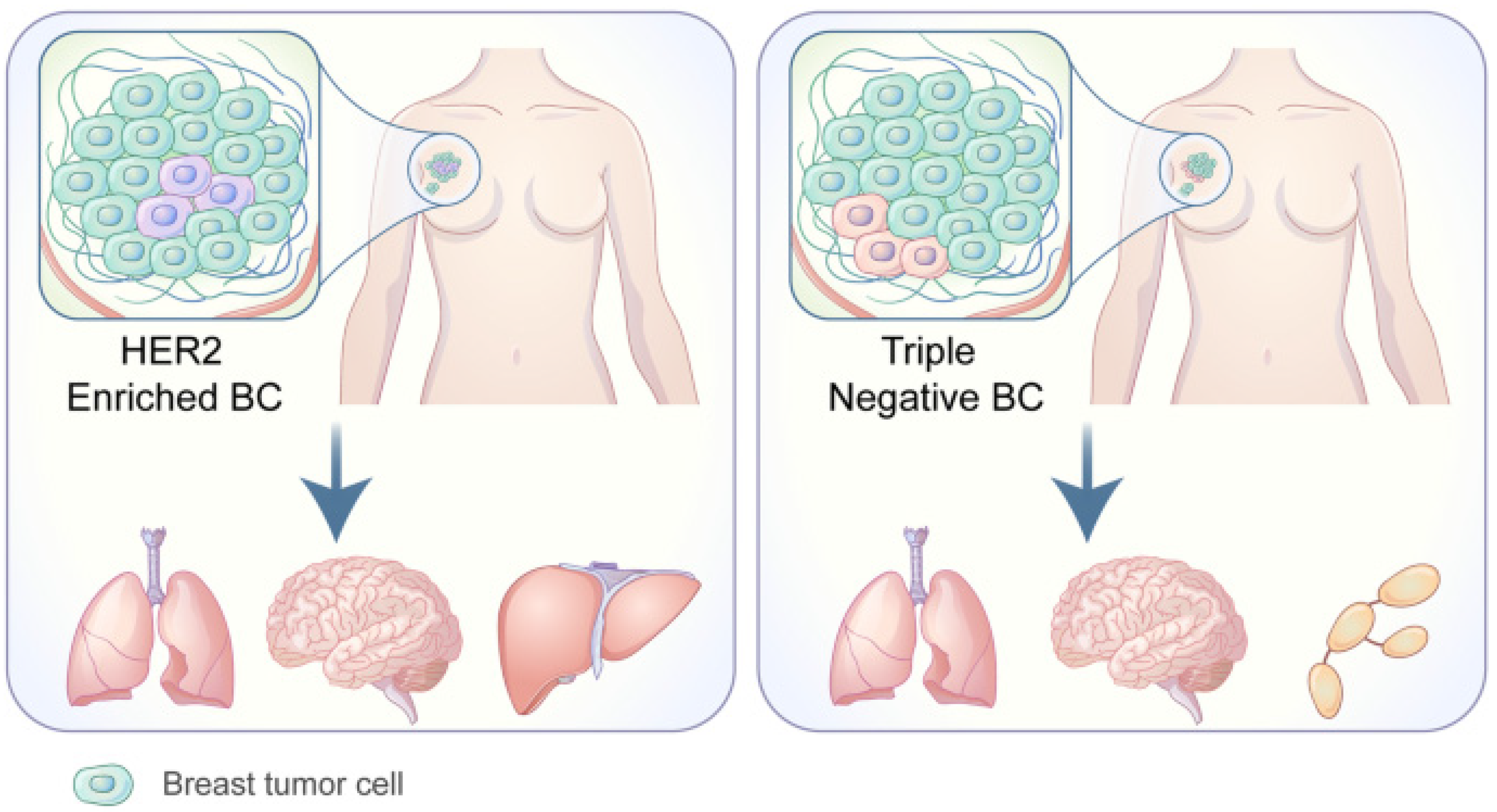
1.1. Organotropism: Breast Cancer Molecular Subtypes
1.2. Clinical Presentation and Detection of Breast Cancer
1.3. Clinical Implications: Successes of Targeted Therapy
2. Role of Genetics and Genomics in Primary Breast Cancer
2.1. Overview
2.2. Other Genes of Interest
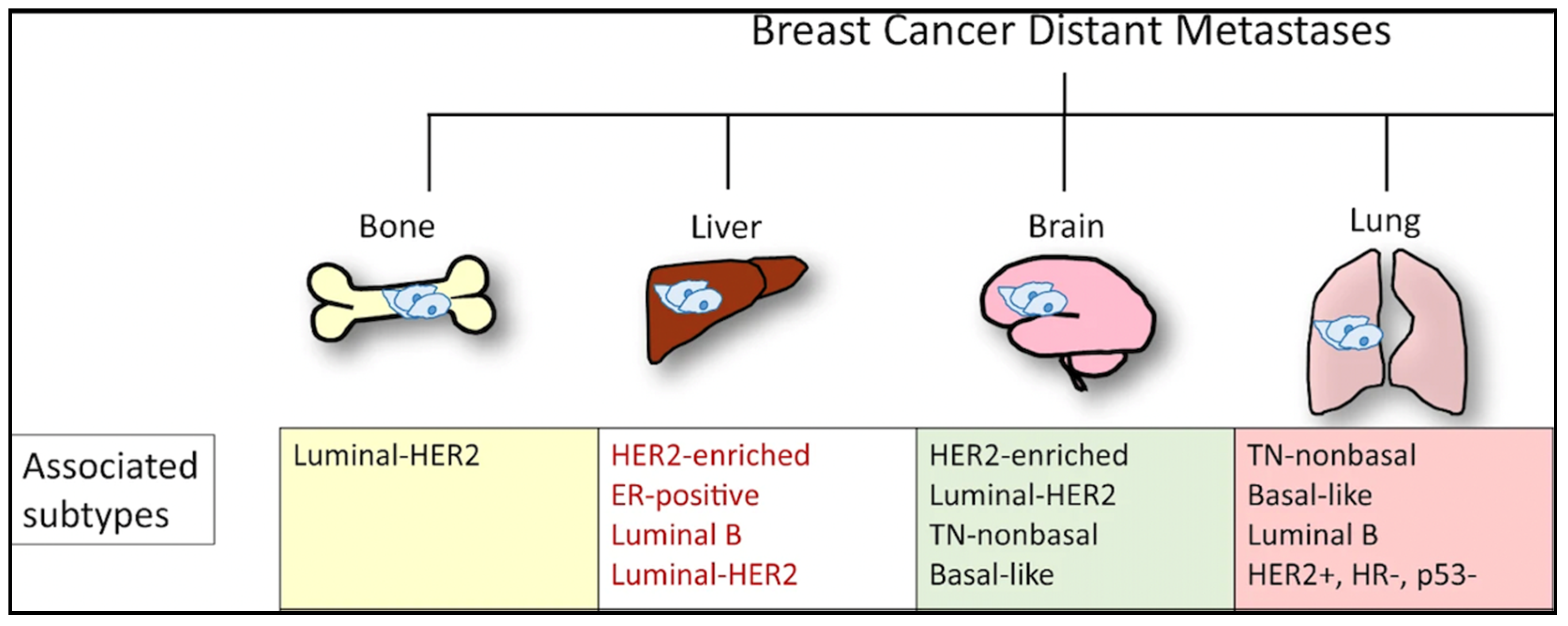
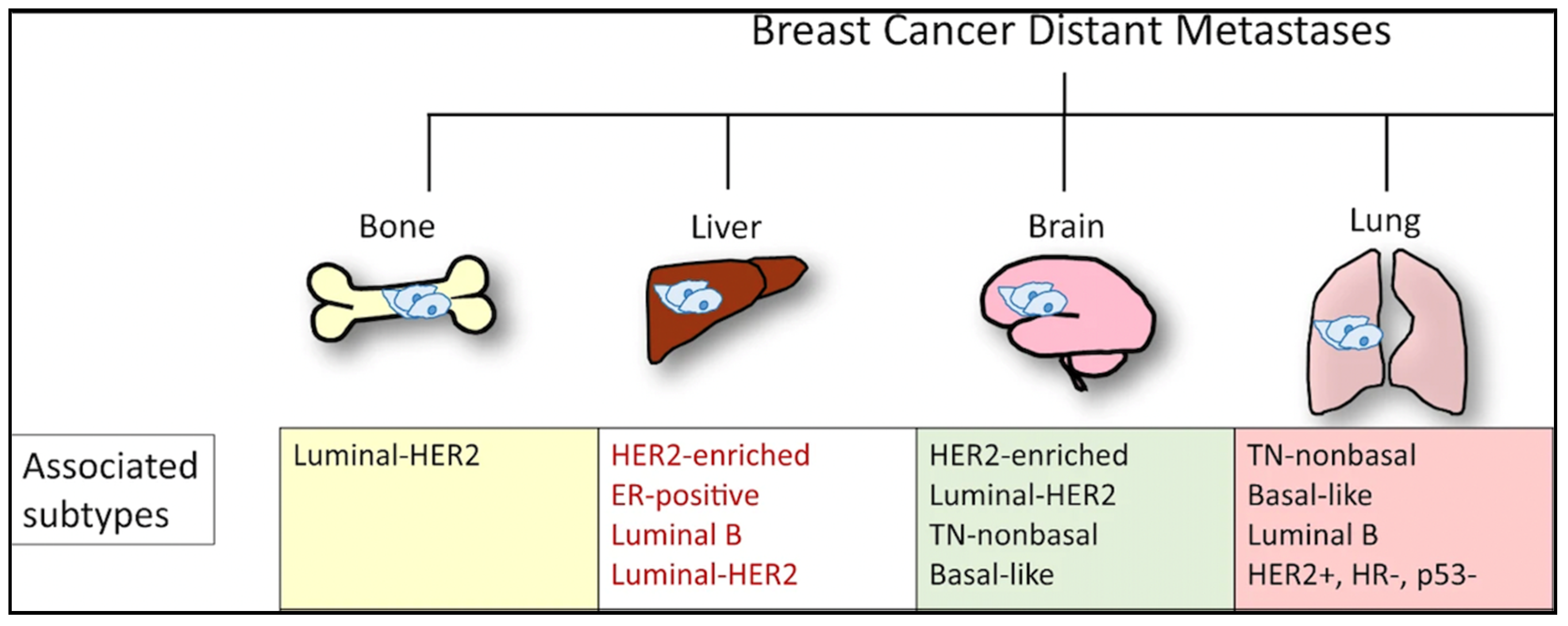
3. Genomics of Metastasis: Most Common Sites
3.1. Bone Metastasis
3.2. Lung Metastasis
3.3. Liver Metastasis
3.4. Brain Metastasis
4. Genomics of Breast Cancer Metastasis: Selected Rare Sites
4.1. Orbital Metastasis
4.2. Gynecologic Metastasis
4.3. Pancreatic Metastasis
5. Paired Primary and Metastatic Breast Cancer
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.M.; Webb-Vargas, Y.; Wheeler, W.; Gail, M.H. Proportion of U.S. Trends in Breast Cancer Incidence Attributable to Long-term Changes in Risk Factor Distributions. Cancer Epidemiol. Biomark. Prev. 2018, 27, 1214–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, C.; Pompei, F.; Burmistrov, D.; Welch, H.G.; Abebe, R.; Wilson, R. Breast Cancer Screening, Incidence, and Mortality Across US Counties. JAMA Intern. Med. 2015, 175, 1483–1489. [Google Scholar] [CrossRef] [Green Version]
- Bleyer, A.; Welch, H.G. Effect of three decades of screening mammography on breast-cancer incidence. N. Engl. J. Med. 2012, 367, 1998–2005. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, S.W. Breast cancer screening: A 35-year perspective. Epidemiol. Rev. 2011, 33, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Bombonati, A.; Sgroi, D.C. The molecular pathology of breast cancer progression. J. Pathol. 2011, 223, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Zhang, J.; Beeraka, N.M.; Tang, C.; Babayeva, Y.V.; Sinelnikov, M.Y.; Zhang, X.; Liu, J.; Reshetov, I.V.; Sukocheva, O.A. Advances in prevention and treatment of obesity-driven effects in breast cancers. Front. Oncol. 2021, 2663. [Google Scholar]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2020, 83, 556–569. [Google Scholar] [CrossRef]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Liu, L.; Hao, X.; Song, Z.; Zhi, X.; Zhang, S.; Zhang, J. Correlation between family history and characteristics of breast cancer. Sci. Rep. 2021, 11, 6360. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Nathanson, K.L.; Offit, K. Two decades after BRCA: Setting paradigms in personalized cancer care and prevention. Science 2014, 343, 1466–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Van Loo, P.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H.; et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 2015, 21, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Low, S.K.; Zembutsu, H.; Nakamura, Y. Breast cancer: The translation of big genomic data to cancer precision medicine. Cancer Sci. 2018, 109, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Luen, S.; Virassamy, B.; Savas, P.; Salgado, R.; Loi, S. The genomic landscape of breast cancer and its interaction with host immunity. Breast 2016, 29, 241–250. [Google Scholar] [CrossRef]
- Wu, Q.; Li, J.; Zhu, S.; Wu, J.; Chen, C.; Liu, Q.; Wei, W.; Zhang, Y.; Sun, S. Breast cancer subtypes predict the preferential site of distant metastases: A SEER based study. Oncotarget 2017, 8, 27990–27996. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Santurro, L.; Gasparri, M.L.; Zuber, V.; Fiacco, E.; Gazzetta, G.; Smart, C.E.; Valentini, A.; Gentilini, O.D. Rare sites of breast cancer metastasis: A review. Transl. Cancer Res. 2019, 8, S518–S552. [Google Scholar] [CrossRef]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
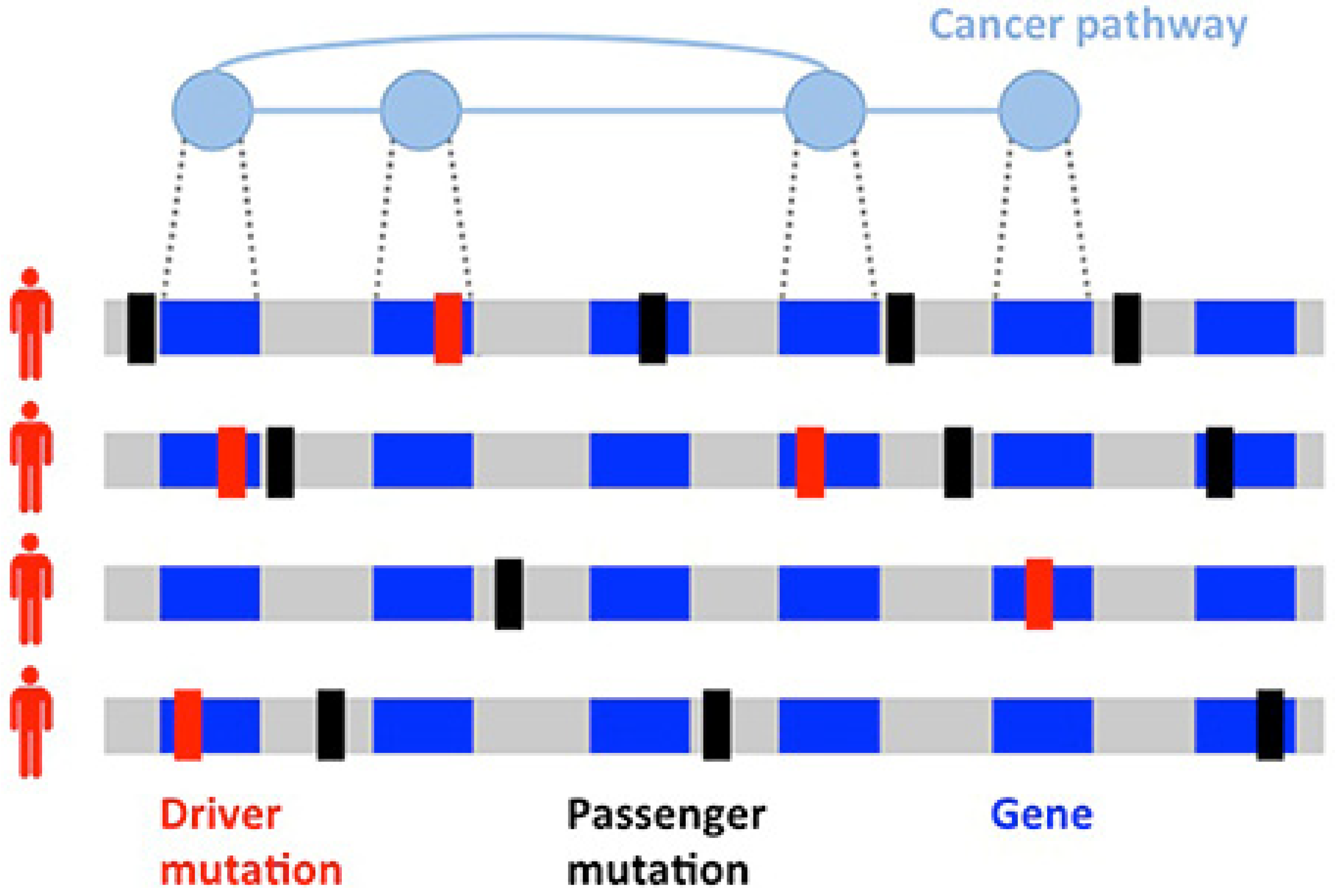
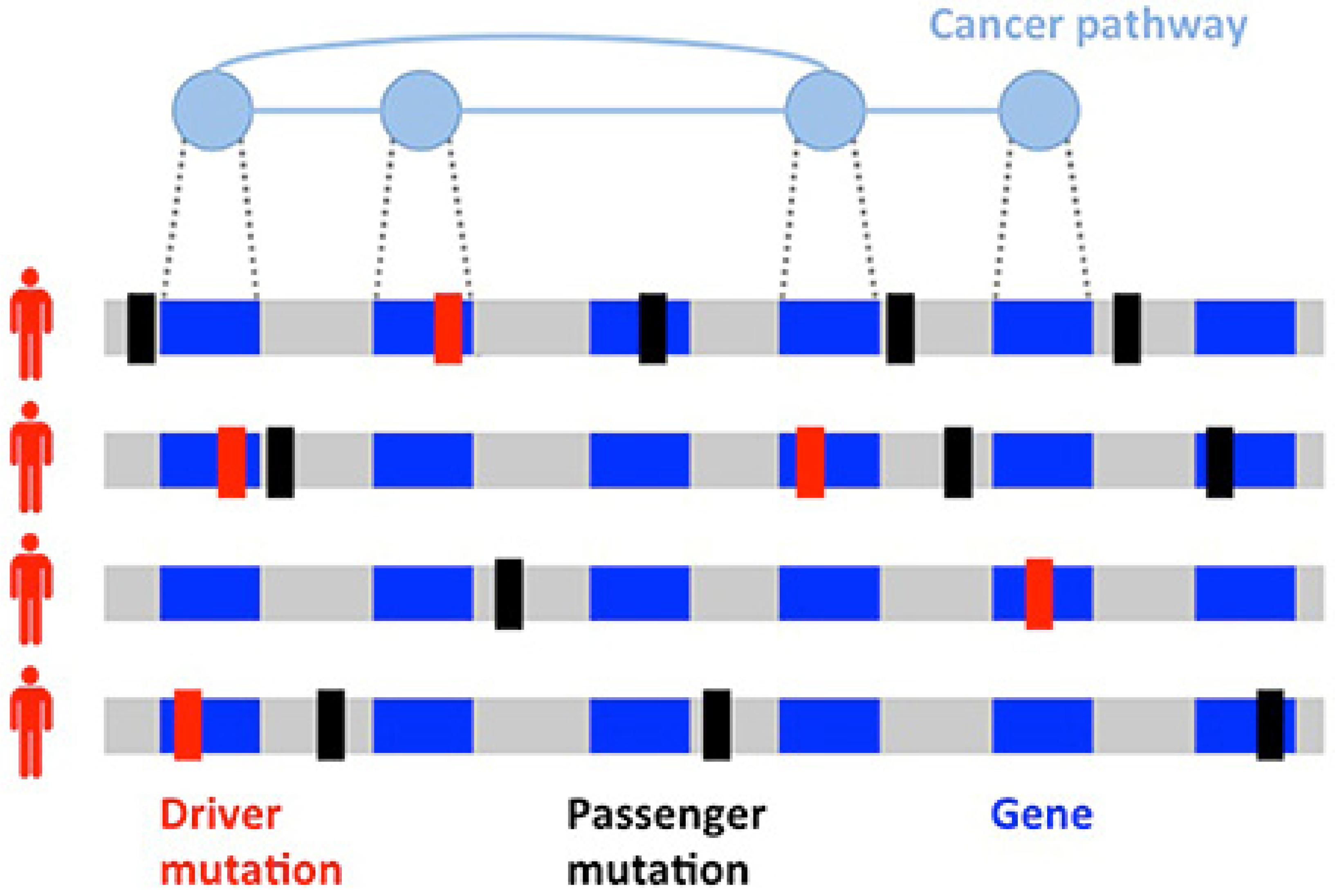
- Vandin, F. Computational methods for characterizing cancer mutational heterogeneity. Front. Genet. 2017, 8, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xu, K.; Wang, R.; Han, X.; Tang, J.; Guan, X. Heterogeneity of BCSCs contributes to the metastatic organotropism of breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 370. [Google Scholar] [CrossRef]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.; Allan, A.L. Molecular Mechanisms of Breast Cancer Metastasis to the Lung: Clinical and Experimental Perspectives. Int. J. Mol. Sci. 2019, 20, 2272. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Hoffmann, A.D.; Liu, H.; Liu, X. Organotropism: New insights into molecular mechanisms of breast cancer metastasis. NPJ Precis. Oncol. 2018, 2, 4. [Google Scholar] [CrossRef]
- Redaniel, M.T.; Martin, R.M.; Ridd, M.J.; Wade, J.; Jeffreys, M. Diagnostic intervals and its association with breast, prostate, lung and colorectal cancer survival in England: Historical cohort study using the Clinical Practice Research Datalink. PLoS ONE 2015, 10, e0126608. [Google Scholar] [CrossRef] [Green Version]
- Koo, M.M.; von Wagner, C.; Abel, G.A.; McPhail, S.; Rubin, G.P.; Lyratzopoulos, G. Typical and atypical presenting symptoms of breast cancer and their associations with diagnostic intervals: Evidence from a national audit of cancer diagnosis. Cancer Epidemiol. 2017, 48, 140–146. [Google Scholar] [CrossRef] [Green Version]
- NICE. 2021 Exceptional Surveillance of Suspected Cancer: Recognition and Referral (NICE Guideline NG12); NICE: London, UK, 2021. [Google Scholar]
- Mook, S.; Van’t Veer, L.J.; Rutgers, E.J.; Ravdin, P.M.; van de Velde, A.O.; van Leeuwen, F.E.; Visser, O.; Schmidt, M.K. Independent prognostic value of screen detection in invasive breast cancer. J. Natl. Cancer Inst. 2011, 103, 585–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, G.C.; Greenberg, D.C.; Britton, P.D.; Chou, P.; Brown, C.H.; Purushotham, A.D.; Duffy, S.W. Screen-detected vs symptomatic breast cancer: Is improved survival due to stage migration alone? Br. J. Cancer 2008, 98, 1741–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irvin, V.L.; Zhang, Z.; Simon, M.S.; Chlebowski, R.T.; Luoh, S.W.; Shadyab, A.H.; Krok-Schoen, J.L.; Tabung, F.K.; Qi, L.; Stefanick, M.L.; et al. Comparison of Mortality Among Participants of Women’s Health Initiative Trials With Screening-Detected Breast Cancers vs Interval Breast Cancers. JAMA Netw. Open 2020, 3, e207227. [Google Scholar] [CrossRef]
- Farshid, G.; Walters, D. Molecular subtypes of screen-detected breast cancer. Breast Cancer Res. Treat. 2018, 172, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [Green Version]
- Collett, K.; Stefansson, I.M.; Eide, J.; Braaten, A.; Wang, H.; Eide, G.E.; Thoresen, S.O.; Foulkes, W.D.; Akslen, L.A. A basal epithelial phenotype is more frequent in interval breast cancers compared with screen detected tumors. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1108–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirsh, V.A.; Chiarelli, A.M.; Edwards, S.A.; O’Malley, F.P.; Shumak, R.S.; Yaffe, M.J.; Boyd, N.F. Tumor characteristics associated with mammographic detection of breast cancer in the Ontario breast screening program. J. Natl. Cancer Inst. 2011, 103, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Unni, N.; Peng, Y. The Changing Paradigm for the Treatment of HER2-Positive Breast Cancer. Cancers 2020, 12, 2081. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Martinez-Saez, O.; Prat, A. Current and Future Management of HER2-Positive Metastatic Breast Cancer. JCO Oncol. Pract. 2021, 17, 594–604. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- van Geelen, C.T.; Savas, P.; Teo, Z.L.; Luen, S.J.; Weng, C.F.; Ko, Y.A.; Kuykhoven, K.S.; Caramia, F.; Salgado, R.; Francis, P.A.; et al. Clinical implications of prospective genomic profiling of metastatic breast cancer patients. Breast Cancer Res. 2020, 22, 91. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.S.; et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Final overall survival results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermida-Prado, F.; Jeselsohn, R. The ESR1 Mutations: From Bedside to Bench to Bedside. Cancer Res. 2021, 81, 537–538. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Boer, K.; Bondarenko, I.; Patel, R.; Pinter, T.; Schmidt, M.; Shparyk, Y.V.; Thummala, A.; Voitko, N.; Bananis, E.; et al. Overall survival results from the randomized phase 2 study of palbociclib in combination with letrozole versus letrozole alone for first-line treatment of ER+/HER2- advanced breast cancer (PALOMA-1, TRIO-18). Breast Cancer Res. Treat. 2020, 183, 419–428. [Google Scholar] [CrossRef]
- Rugo, H.S.; Finn, R.S.; Dieras, V.; Ettl, J.; Lipatov, O.; Joy, A.A.; Harbeck, N.; Castrellon, A.; Iyer, S.; Lu, D.R.; et al. Palbociclib plus letrozole as first-line therapy in estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer with extended follow-up. Breast Cancer Res. Treat. 2019, 174, 719–729. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.C.; Ro, J.; Andre, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Hart, L.; Campone, M.; Petrakova, K.; Winer, E.P.; Janni, W.; et al. Overall Survival with Ribociclib plus Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2022, 386, 942–950. [Google Scholar] [CrossRef]
- Johnston, S.; Martin, M.; Di Leo, A.; Im, S.A.; Awada, A.; Forrester, T.; Frenzel, M.; Hardebeck, M.C.; Cox, J.; Barriga, S.; et al. MONARCH 3 final PFS: A randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 2019, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 mutations in breast cancer. Cancer 2019, 125, 3714–3728. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Goessl, C.; Domchek, S. Olaparib for Metastatic Germline BRCA-Mutated Breast Cancer. N. Engl. J. Med. 2017, 377, 1792–1793. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Garber, J.E. PARP inhibition in breast cancer: Progress made and future hopes. Npj Breast Cancer 2022, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, M.C.; Mitchell, C.A. Phosphoinositide 3-kinase and INPP4B in human breast cancer. Ann. N. Y. Acad. Sci. 2013, 1280, 1–5. [Google Scholar] [CrossRef]
- Rodgers, S.J.; Ooms, L.M.; Oorschot, V.M.J.; Schittenhelm, R.B.; Nguyen, E.V.; Hamila, S.A.; Rynkiewicz, N.; Gurung, R.; Eramo, M.J.; Sriratana, A.; et al. INPP4B promotes PI3Kalpha-dependent late endosome formation and Wnt/beta-catenin signaling in breast cancer. Nat. Commun. 2021, 12, 3140. [Google Scholar] [CrossRef]
- Watt, L.F.; Panicker, N.; Mannan, A.; Copeland, B.; Kahl, R.G.S.; Dun, M.D.; Young, B.; Roselli, S.; Verrills, N.M. Functional importance of PP2A regulatory subunit loss in breast cancer. Breast Cancer Res. Treat. 2017, 166, 117–131. [Google Scholar] [CrossRef]
- Bertino, J.R.; Waud, W.R.; Parker, W.B.; Lubin, M. Targeting tumors that lack methylthioadenosine phosphorylase (MTAP) activity: Current strategies. Cancer Biol. Ther. 2011, 11, 627–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalev, P.; Hyer, M.L.; Gross, S.; Konteatis, Z.; Chen, C.C.; Fletcher, M.; Lein, M.; Aguado-Fraile, E.; Frank, V.; Barnett, A.; et al. MAT2A Inhibition Blocks the Growth of MTAP-Deleted Cancer Cells by Reducing PRMT5-Dependent mRNA Splicing and Inducing DNA Damage. Cancer Cell 2021, 39, 209–224.e211. [Google Scholar] [CrossRef]
- Konteatis, Z.; Travins, J.; Gross, S.; Marjon, K.; Barnett, A.; Mandley, E.; Nicolay, B.; Nagaraja, R.; Chen, Y.; Sun, Y.; et al. Discovery of AG-270, a First-in-Class Oral MAT2A Inhibitor for the Treatment of Tumors with Homozygous MTAP Deletion. J. Med. Chem. 2021, 64, 4430–4449. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Nixon, M.J.; Formisano, L.; Mayer, I.A.; Estrada, M.V.; Gonzalez-Ericsson, P.I.; Isakoff, S.J.; Forero-Torres, A.; Won, H.; Sanders, M.E.; Solit, D.B.; et al. PIK3CA and MAP3K1 alterations imply luminal A status and are associated with clinical benefit from pan-PI3K inhibitor buparlisib and letrozole in ER+ metastatic breast cancer. NPJ Breast Cancer 2019, 5, 31. [Google Scholar] [CrossRef]
- Avivar-Valderas, A.; McEwen, R.; Taheri-Ghahfarokhi, A.; Carnevalli, L.S.; Hardaker, E.L.; Maresca, M.; Hudson, K.; Harrington, E.A.; Cruzalegui, F. Functional significance of co-occurring mutations in PIK3CA and MAP3K1 in breast cancer. Oncotarget 2018, 9, 21444–21458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Zhou, S.; Yang, Y.; Xu, Y.; Gong, X.; Cheng, Y.; Wang, Y. LncRNA MNX1-AS1: A novel oncogenic propellant in cancers. Biomed. Pharmacother. 2022, 149, 112801. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Wang, M.; Zhu, Y.; Zhu, W.; Yang, T.; Li, H.; Lin, S.; Dai, C.; Deng, Y.; Song, D.; et al. Expression, Clinical Significance, and Functional Prediction of MNX1 in Breast Cancer. Mol. Ther. Nucleic Acids 2018, 13, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, Q.; Li, D.; Shen, Z.; Zhang, K.; Bi, Z.; Li, Y. Long Non-Coding RNA MNX1-AS1 Promotes Progression of Triple Negative Breast Cancer by Enhancing Phosphorylation of Stat3. Front. Oncol. 2020, 10, 1108. [Google Scholar] [CrossRef]
- Soni, A.; Ren, Z.; Hameed, O.; Chanda, D.; Morgan, C.J.; Siegal, G.P.; Wei, S. Breast cancer subtypes predispose the site of distant metastases. Am. J. Clin. Pathol. 2015, 143, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Smid, M.; Wang, Y.; Klijn, J.G.M.; Sieuwerts, A.M.; Zhang, Y.; Atkins, D.; Martens, J.W.M.; Foekens, J.A. Genes Associated With Breast Cancer Metastatic to Bone. J. Clin. Oncol. 2006, 24, 2261–2267. [Google Scholar] [CrossRef] [PubMed]
- Spadazzi, C.; Mercatali, L.; Esposito, M.; Wei, Y.; Liverani, C.; De Vita, A.; Miserocchi, G.; Carretta, E.; Zanoni, M.; Cocchi, C.; et al. Trefoil factor-1 upregulation in estrogen-receptor positive breast cancer correlates with an increased risk of bone metastasis. Bone 2021, 144, 115775. [Google Scholar] [CrossRef] [PubMed]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer (Review). Front. Med. 2017, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cabioglu, N.; Sahin, A.A.; Morandi, P.; Meric-Bernstam, F.; Islam, R.; Lin, H.Y.; Bucana, C.D.; Gonzalez-Angulo, A.M.; Hortobagyi, G.N.; Cristofanilli, M. Chemokine receptors in advanced breast cancer: Differential expression in metastatic disease sites with diagnostic and therapeutic implications. Ann. Oncol. 2009, 20, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Sihto, H.; Lundin, J.; Lundin, M.; Lehtimaki, T.; Ristimaki, A.; Holli, K.; Sailas, L.; Kataja, V.; Turpeenniemi-Hujanen, T.; Isola, J.; et al. Breast cancer biological subtypes and protein expression predict for the preferential distant metastasis sites: A nationwide cohort study. Breast Cancer Res. 2011, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef]
- Kono, M.; Fujii, T.; Matsuda, N.; Harano, K.; Chen, H.; Wathoo, C.; Joon, A.Y.; Tripathy, D.; Meric-Bernstam, F.; Ueno, N.T. Somatic mutations, clinicopathologic characteristics, and survival in patients with untreated breast cancer with bone-only and non-bone sites of first metastasis. J. Cancer 2018, 9, 3640–3646. [Google Scholar] [CrossRef]
- Yousefi, M.; Nosrati, R.; Salmaninejad, A.; Dehghani, S.; Shahryari, A.; Saberi, A. Organ-specific metastasis of breast cancer: Molecular and cellular mechanisms underlying lung metastasis. Cell Oncol. 2018, 41, 123–140. [Google Scholar] [CrossRef]
- Jin, L.; Han, B.; Siegel, E.; Cui, Y.; Giuliano, A.; Cui, X. Breast cancer lung metastasis: Molecular biology and therapeutic implications. Cancer Biol. Ther. 2018, 19, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef]
- Higa, A.; Mulot, A.; Delom, F.; Bouchecareilh, M.; Nguyen, D.T.; Boismenu, D.; Wise, M.J.; Chevet, E. Role of pro-oncogenic protein disulfide isomerase (PDI) family member anterior gradient 2 (AGR2) in the control of endoplasmic reticulum homeostasis. J. Biol. Chem. 2011, 286, 44855–44868. [Google Scholar] [CrossRef] [Green Version]
- Salmans, M.L.; Zhao, F.; Andersen, B. The estrogen-regulated anterior gradient 2 (AGR2) protein in breast cancer: A potential drug target and biomarker. Breast Cancer Res. 2013, 15, 204. [Google Scholar] [CrossRef] [Green Version]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Qin, J.; Zhou, Z.; Chen, W.; Wang, C.; Zhang, H.; Ge, G.; Shao, M.; You, D.; Fan, Z.; Xia, H.; et al. BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nat. Commun. 2015, 6, 8471. [Google Scholar] [CrossRef]
- Jia, L.; Zhou, Z.; Liang, H.; Wu, J.; Shi, P.; Li, F.; Wang, Z.; Wang, C.; Chen, W.; Zhang, H.; et al. KLF5 promotes breast cancer proliferation, migration and invasion in part by upregulating the transcription of TNFAIP2. Oncogene 2016, 35, 2040–2051. [Google Scholar] [CrossRef]
- Zheng, H.Q.; Zhou, Z.; Huang, J.; Chaudhury, L.; Dong, J.T.; Chen, C. Kruppel-like factor 5 promotes breast cell proliferation partially through upregulating the transcription of fibroblast growth factor binding protein 1. Oncogene 2009, 28, 3702–3713. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Wang, C.; Chen, W.; Zhang, H.; Chaudhury, L.; Zhou, Z.; Liu, R.; Chen, C. Kruppel-like factor 5 transcription factor promotes microsomal prostaglandin E2 synthase 1 gene transcription in breast cancer. J. Biol. Chem. 2013, 288, 26731–26740. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Nie, Z.; Zhou, Z.; Zhang, H.; Liu, R.; Wu, J.; Qin, J.; Ma, Y.; Chen, L.; Li, S.; et al. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27Kip1. Oncotarget 2015, 6, 17685–17697. [Google Scholar] [CrossRef] [Green Version]
- Obenauf, A.C.; Massague, J. Surviving at a Distance: Organ-Specific Metastasis. Trends Cancer 2015, 1, 76–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador, F.; Martin, A.; Lopez-Menendez, C.; Moreno-Bueno, G.; Santos, V.; Vazquez-Naharro, A.; Santamaria, P.G.; Morales, S.; Dubus, P.R.; Muinelo-Romay, L.; et al. Lysyl Oxidase-like Protein LOXL2 Promotes Lung Metastasis of Breast Cancer. Cancer Res. 2017, 77, 5846–5859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zheng, Z.; Chen, L.; Zheng, H. MAPK, NFkappaB, and VEGF signaling pathways regulate breast cancer liver metastasis. Oncotarget 2017, 8, 101452–101460. [Google Scholar] [CrossRef] [Green Version]
- Braso-Maristany, F.; Pare, L.; Chic, N.; Martinez-Saez, O.; Pascual, T.; Mallafre-Larrosa, M.; Schettini, F.; Gonzalez-Farre, B.; Sanfeliu, E.; Martinez, D.; et al. Gene expression profiles of breast cancer metastasis according to organ site. Mol. Oncol. 2022, 16, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Liu, S.; Wang, Y.; Song, X. Prognosis and Genomic Landscape of Liver Metastasis in Patients with Breast Cancer. Front. Oncol. 2021, 11, 588136. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, N.N.; Huang, D.J.; Luo, Y.C.; Huang, J.Z.; He, H.Y.; Lu, H.L.; Song, W.L. PPFIA1 is upregulated in liver metastasis of breast cancer and is a potential poor prognostic indicator of metastatic relapse. Tumour Biol. 2017, 39, 1010428317713492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, L.V.; Tate, C.R.; Hoang, V.T.; Burks, H.E.; Gilliam, D.; Martin, E.C.; Elliott, S.; Miller, D.B.; Buechlein, A.; Rusch, D.; et al. Regulation of triple-negative breast cancer cell metastasis by the tumor-suppressor liver kinase B1. Oncogenesis 2015, 4, e168. [Google Scholar] [CrossRef] [Green Version]
- Salhia, B.; Kiefer, J.; Ross, J.T.; Metapally, R.; Martinez, R.A.; Johnson, K.N.; DiPerna, D.M.; Paquette, K.M.; Jung, S.; Nasser, S.; et al. Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PLoS ONE 2014, 9, e85448. [Google Scholar] [CrossRef]
- Weil, R.J.; Palmieri, D.C.; Bronder, J.L.; Stark, A.M.; Steeg, P.S. Breast cancer metastasis to the central nervous system. Am. J. Pathol. 2005, 167, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.S.; Huang, S.; Lu, W.; Lev, D.C.; Price, J.E. Vascular endothelial growth factor expression promotes the growth of breast cancer brain metastases in nude mice. Clin. Exp. Metastasis 2004, 21, 107–118. [Google Scholar] [CrossRef]
- Raeder, M.B.; Birkeland, E.; Trovik, J.; Krakstad, C.; Shehata, S.; Schumacher, S.; Zack, T.I.; Krohn, A.; Werner, H.M.; Moody, S.E.; et al. Integrated genomic analysis of the 8q24 amplification in endometrial cancers identifies ATAD2 as essential to MYC-dependent cancers. PLoS ONE 2013, 8, e54873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciro, M.; Prosperini, E.; Quarto, M.; Grazini, U.; Walfridsson, J.; McBlane, F.; Nucifero, P.; Pacchiana, G.; Capra, M.; Christensen, J.; et al. ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res. 2009, 69, 8491–8498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hua, H.; Ran, Y.; Zhang, H.; Liu, W.; Yang, Z.; Jiang, Y. Derlin-1 is overexpressed in human breast carcinoma and protects cancer cells from endoplasmic reticulum stress-induced apoptosis. Breast Cancer Res. 2008, 10, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Li, W.; Liu, N.; Zhang, F.; Liu, H.; Liu, F.; Liu, J.; Zhang, T.; Niu, Y. Nek2A contributes to tumorigenic growth and possibly functions as potential therapeutic target for human breast cancer. J. Cell Biochem. 2012, 113, 1904–1914. [Google Scholar] [CrossRef] [PubMed]
- Baysal, B.E.; Willett-Brozick, J.E.; Taschner, P.E.; Dauwerse, J.G.; Devilee, P.; Devlin, B. A high-resolution integrated map spanning the SDHD gene at 11q23: A 1.1-Mb BAC contig, a partial transcript map and 15 new repeat polymorphisms in a tumour-suppressor region. Eur. J. Hum. Genet. 2001, 9, 121–129. [Google Scholar] [CrossRef]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef]
- Mehrotra, J.; Vali, M.; McVeigh, M.; Kominsky, S.L.; Fackler, M.J.; Lahti-Domenici, J.; Polyak, K.; Sacchi, N.; Garrett-Mayer, E.; Argani, P.; et al. Very high frequency of hypermethylated genes in breast cancer metastasis to the bone, brain, and lung. Clin. Cancer Res. 2004, 10, 3104–3109. [Google Scholar] [CrossRef] [Green Version]
- Bonavolonta, G.; Strianese, D.; Grassi, P.; Comune, C.; Tranfa, F.; Uccello, G.; Iuliano, A. An analysis of 2480 space-occupying lesions of the orbit from 1976 to 2011. Ophthalmic. Plast. Reconstr. Surg. 2013, 29, 79–86. [Google Scholar] [CrossRef]
- Ahmad, S.M.; Esmaeli, B. Metastatic tumors of the orbit and ocular adnexa. Curr. Opin. Ophthalmol. 2007, 18, 405–413. [Google Scholar] [CrossRef]
- Shields, J.A.; Shields, C.L.; Scartozzi, R. Survey of 1264 patients with orbital tumors and simulating lesions: The 2002 Montgomery Lecture, part 1. Ophthalmology 2004, 111, 997–1008. [Google Scholar] [CrossRef]
- Shields, J.A.; Shields, C.L.; Brotman, H.K.; Carvalho, C.; Perez, N.; Eagle, R.C., Jr. Cancer metastatic to the orbit: The 2000 Robert M. Curts Lecture. Ophthalmic. Plast. Reconstr. Surg. 2001, 17, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, A.A.; Archibald, C.W.; Fleming, B.; Ong, L.; O’Donnell, B.; Crompton, J.J.; Selva, D.; McNab, A.A.; Sullivan, T.J. Orbital metastasis: Clinical features, management and outcome. Orbit 2009, 28, 153–159. [Google Scholar] [CrossRef]
- Raap, M.; Antonopoulos, W.; Dammrich, M.; Christgen, H.; Steinmann, D.; Langer, F.; Lehmann, U.; Kreipe, H.; Christgen, M. High frequency of lobular breast cancer in distant metastases to the orbit. Cancer Med. 2015, 4, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; El-Sayed, M.E.; Powe, D.G.; Green, A.R.; Habashy, H.; Grainge, M.J.; Robertson, J.F.; Blamey, R.; Gee, J.; Nicholson, R.I.; et al. Invasive lobular carcinoma of the breast: Response to hormonal therapy and outcomes. Eur. J. Cancer 2008, 44, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Spelsberg, H.; Klueppel, M.; Reinhard, T.; Glaeser, M.; Niederacher, D.; Beckmann, M.W.; Sundmacher, R. Detection of oestrogen receptors (ER) alpha and beta in conjunctiva, lacrimal gland, and tarsal plates. Eye 2004, 18, 729–733. [Google Scholar] [CrossRef]
- Grajales-Alvarez, R.; Gutierrez-Mata, A. Orbital metastases from breast cancer: A retrospective analysis of 28 cases. Cancer Treat. Res. Commun. 2020, 24, 100184. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.M.; Tebit, E.V.; El Sayed, A.; Smolkin, M.E.; Dillon, P.M. Orbital Metastases from Breast Cancer: Retrospective Analysis at an Academic Cancer Center. Breast J. 2016, 22, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Kutasovic, J.R.; McCart Reed, A.E.; Males, R.; Sim, S.; Saunus, J.M.; Dalley, A.; McEvoy, C.R.; Dedina, L.; Miller, G.; Peyton, S.; et al. Breast cancer metastasis to gynaecological organs: A clinico-pathological and molecular profiling study. J. Pathol. Clin. Res. 2019, 5, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Drago, J.Z.; Formisano, L.; Juric, D.; Niemierko, A.; Servetto, A.; Wander, S.A.; Spring, L.M.; Vidula, N.; Younger, J.; Peppercorn, J.; et al. FGFR1 Amplification Mediates Endocrine Resistance but Retains TORC Sensitivity in Metastatic Hormone Receptor-Positive (HR(+)) Breast Cancer. Clin. Cancer Res. 2019, 25, 6443–6451. [Google Scholar] [CrossRef] [Green Version]
- Santolla, M.F.; Maggiolini, M. The FGF/FGFR System in Breast Cancer: Oncogenic Features and Therapeutic Perspectives. Cancers 2020, 12, 3029. [Google Scholar] [CrossRef]
- Ballarin, R.; Spaggiari, M.; Cautero, N.; De Ruvo, N.; Montalti, R.; Longo, C.; Pecchi, A.; Giacobazzi, P.; De Marco, G.; D’Amico, G.; et al. Pancreatic metastases from renal cell carcinoma: The state of the art. World J. Gastroenterol. 2011, 17, 4747–4756. [Google Scholar] [CrossRef] [PubMed]
- Shee, K.; Strait, A.M.; Liu, X. Biomarkers to diagnose metastatic breast carcinoma to the pancreas: A case report and update. Diagn. Cytopathol. 2019, 47, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.K.; Li, J.; Jeong, K.J.; Shao, S.; Chen, H.; Tsang, Y.H.; Sengupta, S.; Wang, Z.; Bhavana, V.H.; Tran, R.; et al. Systematic Functional Annotation of Somatic Mutations in Cancer. Cancer Cell 2018, 33, 450–462.e410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, C.K.; Martelotto, L.G.; Gauthier, A.; Wen, H.C.; Piscuoglio, S.; Lim, R.S.; Cowell, C.F.; Wilkerson, P.M.; Wai, P.; Rodrigues, D.N.; et al. Intra-tumor genetic heterogeneity and alternative driver genetic alterations in breast cancers with heterogeneous HER2 gene amplification. Genome Biol. 2015, 16, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagano, M.; Kohsaka, S.; Ueno, T.; Kojima, S.; Saka, K.; Iwase, H.; Kawazu, M.; Mano, H. High-Throughput Functional Evaluation of Variants of Unknown Significance in ERBB2. Clin. Cancer Res. 2018, 24, 5112–5122. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.R.; Pan, T.-c.; Pant, D.K.; Shih, N.N.; Chen, Y.; Harvey, K.L.; Solomon, A.; Lieberman, D.; Morrissette, J.J.; Soucier-Ernst, D. Genomic landscape of metastatic breast cancer identifies preferentially dysregulated pathways and targets. J. Clin. Investig. 2020, 130, 4252–4265. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Mechanism | BC Molecular Subtype Target | Advancement |
|---|---|---|---|
| Trastuzumab | HER2 receptor inhibitor | HER2+ | Improved survival |
| Alpelisib | α-specific PI3K inhibitor | HR+/HER2− metastatic BCs with PIK3CA mutation | IPFS, when combined with fulvestrant |
| Palbociclib | CD4/6 inhibitor | HR+/HER2− | IPFS following metastasis, when combined with endocrine therapy |
| Ribociclib | CD4/6 inhibitor | HR+/HER2− | IPFS following metastasis, when combined with endocrine therapy |
| Abemaciclib | CD4/6 inhibitor | HR+/HER2− | IPFS following metastasis, when combined with endocrine therapy |
| Everolimus | mTOR inhibitor | HR+/HER2− metastatic BC | IPFS following metastasis, when combined with endocrine therapy |
| Olaparib | PARP inhibitor | HER2− metastatic BCs in germline BRCA mutation carriers | IPFS and health-related quality of life |
| Talazoparib | PARP inhibitor | HER2− metastatic BC (in germline BRCA mutation carriers) | IPFS and health-related quality of life |
| Primary Breast Cancer | Bone | Lung | Liver | Brain |
|---|---|---|---|---|
| PIK3CA TP53 ERBB2 FGFR1 CCND MUC16 AHNAK2 SYNE1 KMT2C GATA3 AKT1 PTEN INPP4B PPP2R2A MTAP MAP2K4 MNX1 | TFF1 TFF3 AGR2 NAT1 CR1P1 CXCR4 CCR7 SNAI1 IL11 CTGF CXCR4 MMP-1 | MMP1 MMP2 CXCL1 PTGS2 ID1 VCAM1 EREG SPARC IL13RA2 AGR2 KLF5 Loxl2 | MAPK NFκB VEGF ESR1 AKT1 ERBB2 FGFR4 MS APOBEC cytidine deaminases PPFIA1 | PAM50 VEGF-A IL-8 ATAD2 DERL1 NEK2A ATM CRYAB HSPB2 ANGPT1 KDR ITGAM PIK3CG TEK BRAF BCL2 AURKA AURKB FOXM1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bennett, C.; Carroll, C.; Wright, C.; Awad, B.; Park, J.M.; Farmer, M.; Brown, E.; Heatherly, A.; Woodard, S. Breast Cancer Genomics: Primary and Most Common Metastases. Cancers 2022, 14, 3046. https://doi.org/10.3390/cancers14133046
Bennett C, Carroll C, Wright C, Awad B, Park JM, Farmer M, Brown E, Heatherly A, Woodard S. Breast Cancer Genomics: Primary and Most Common Metastases. Cancers. 2022; 14(13):3046. https://doi.org/10.3390/cancers14133046
Chicago/Turabian StyleBennett, Caroline, Caleb Carroll, Cooper Wright, Barbara Awad, Jeong Mi Park, Meagan Farmer, Elizabeth (Bryce) Brown, Alexis Heatherly, and Stefanie Woodard. 2022. "Breast Cancer Genomics: Primary and Most Common Metastases" Cancers 14, no. 13: 3046. https://doi.org/10.3390/cancers14133046
APA StyleBennett, C., Carroll, C., Wright, C., Awad, B., Park, J. M., Farmer, M., Brown, E., Heatherly, A., & Woodard, S. (2022). Breast Cancer Genomics: Primary and Most Common Metastases. Cancers, 14(13), 3046. https://doi.org/10.3390/cancers14133046