
Endogenous Pancreatic Cancer Cell PD-1 Activates MET and Induces Epithelial-Mesenchymal Transition to Promote Cancer Progression

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Cell Culture
2.3. Ethics Approval and Patient Tumor Acquisition
2.4. Western Blots
2.5. Co-Immunoprecipitation
2.6. Quantitative PCR
2.7. Wound Healing
2.8. Transwell Cell Migration
2.9. Drug Cytotoxicity Assays
2.10. In Vivo Drug Testing
2.11. Immunohistochemical (IHC) Staining
2.12. Statistical Analysis
3. Results
3.1. Identification of PD-1/PD-L1 Activated Pathways
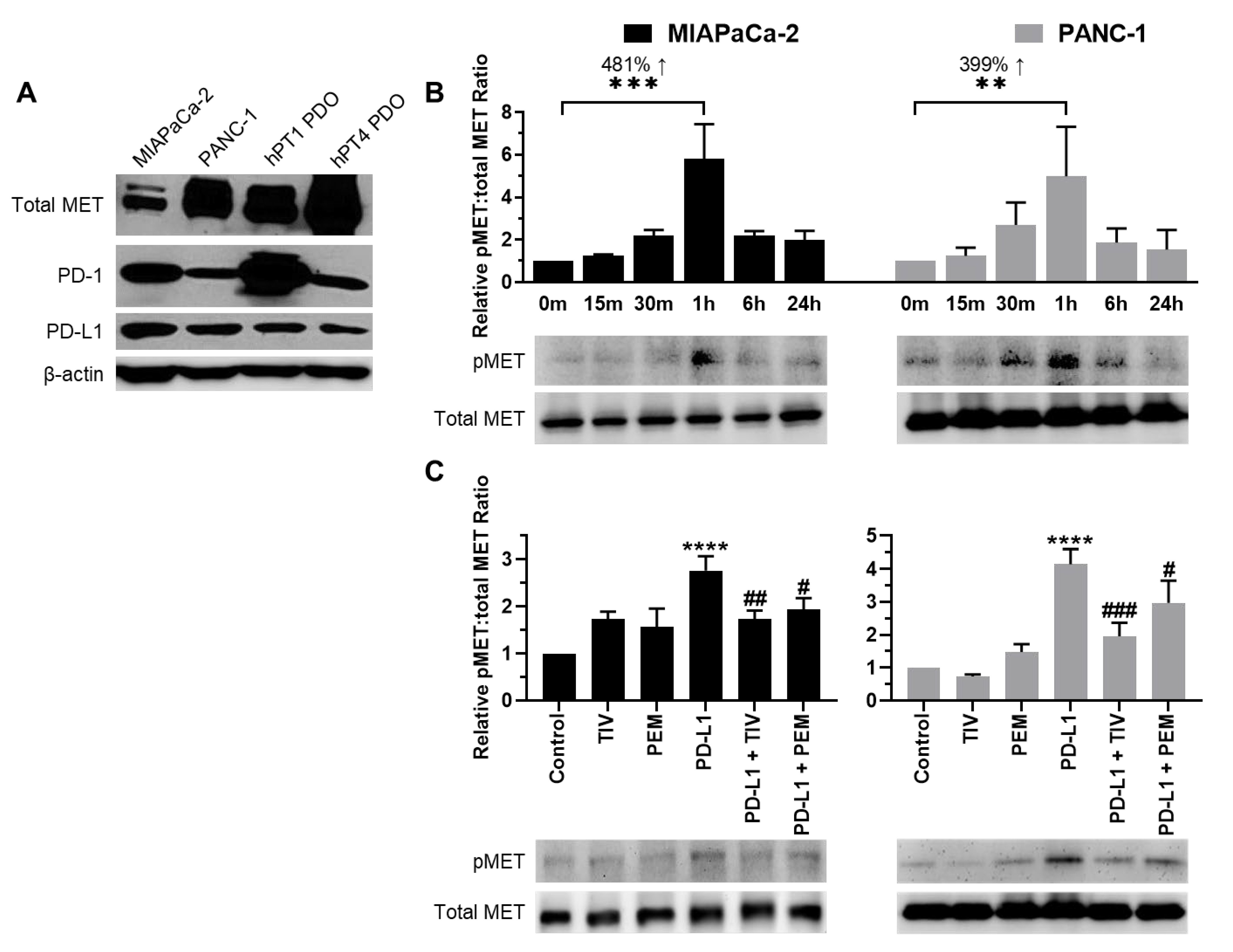
3.2. PD-1 and MET Expression in PDAC Cells and PDOs
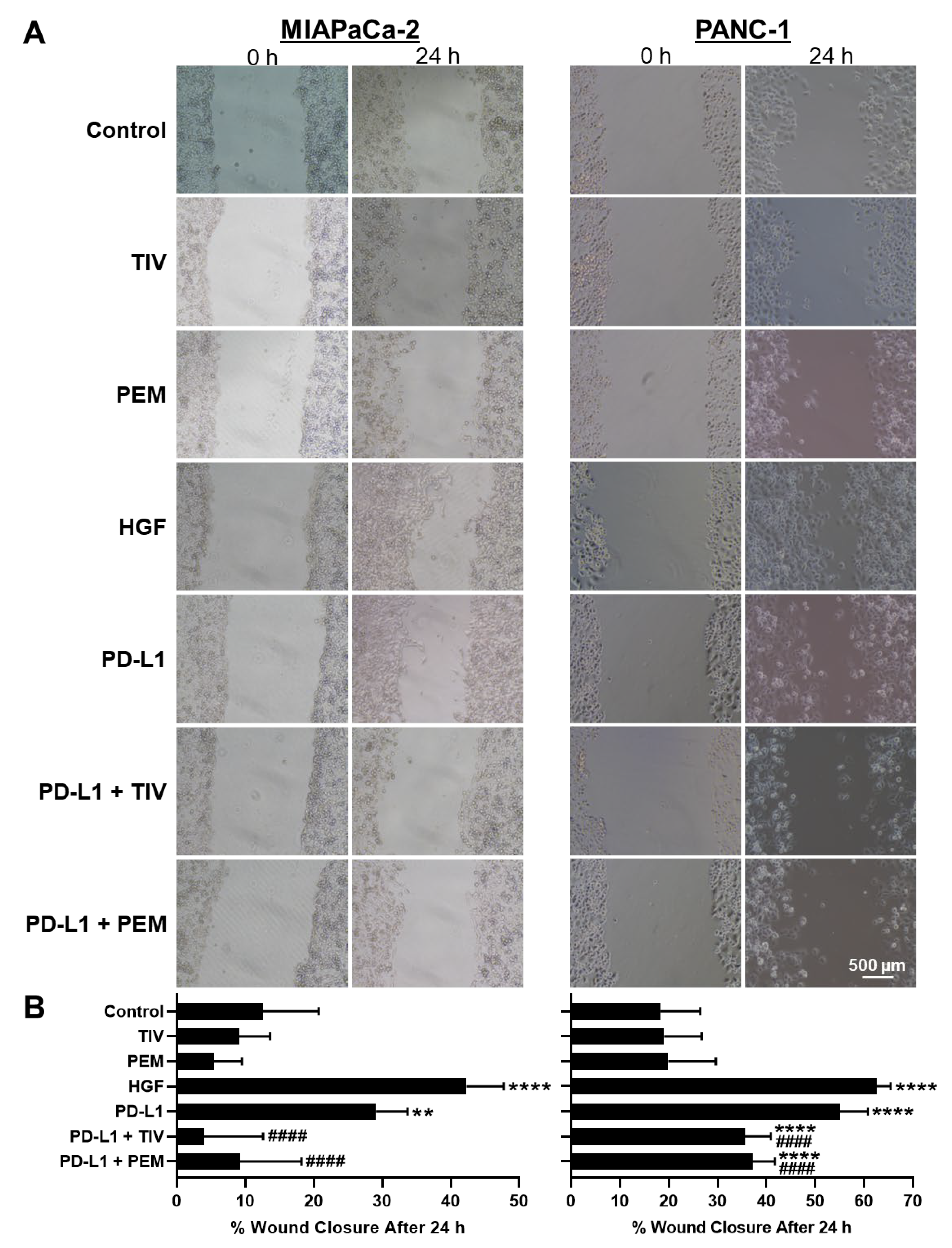
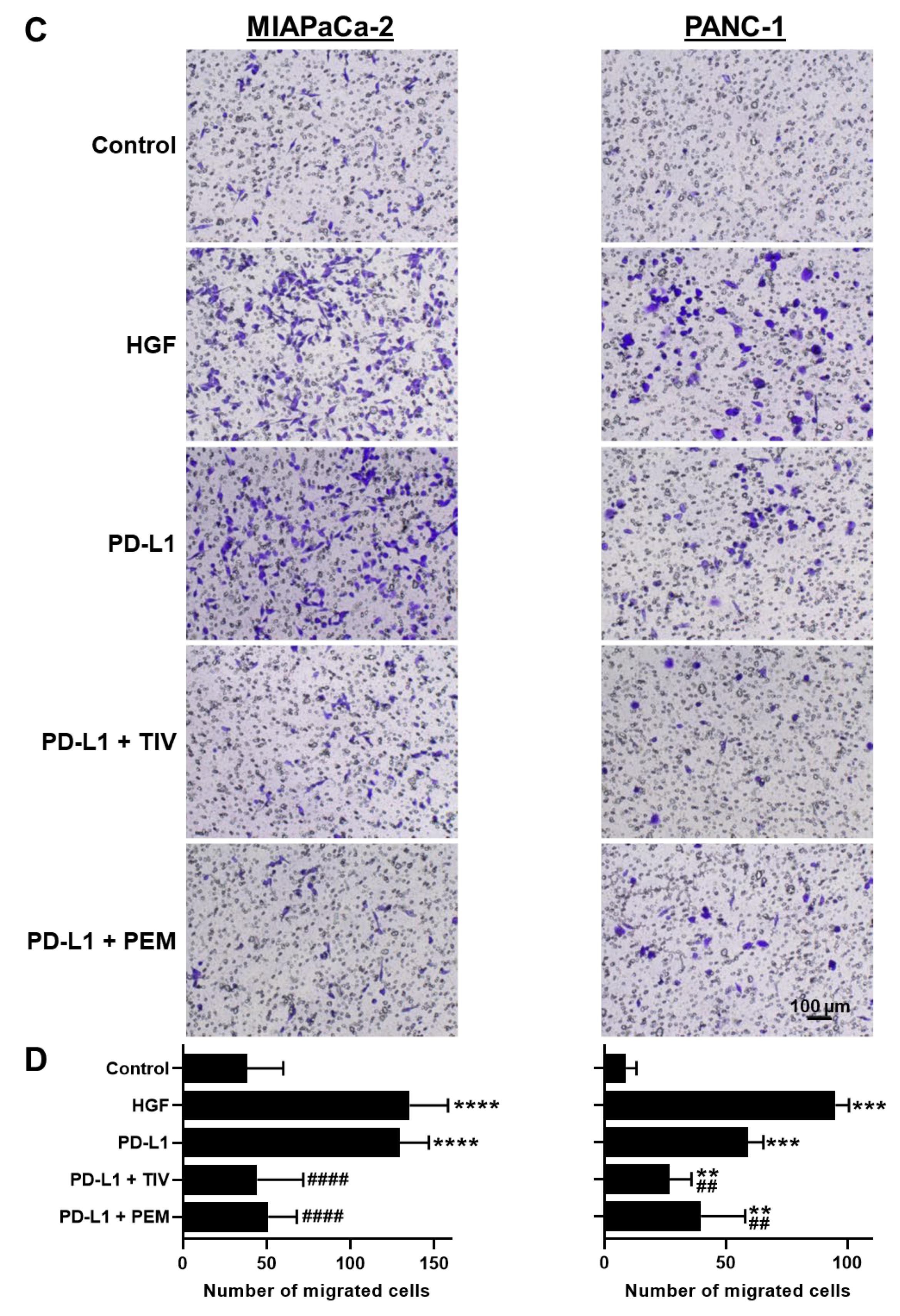
3.3. PD-1 and MET Regulate Cell Motility and Migration
3.4. The PD-1/PD-L1 Axis Induces EMT
3.5. PD-1 and PD-L1 Do Not Directly Interact with MET
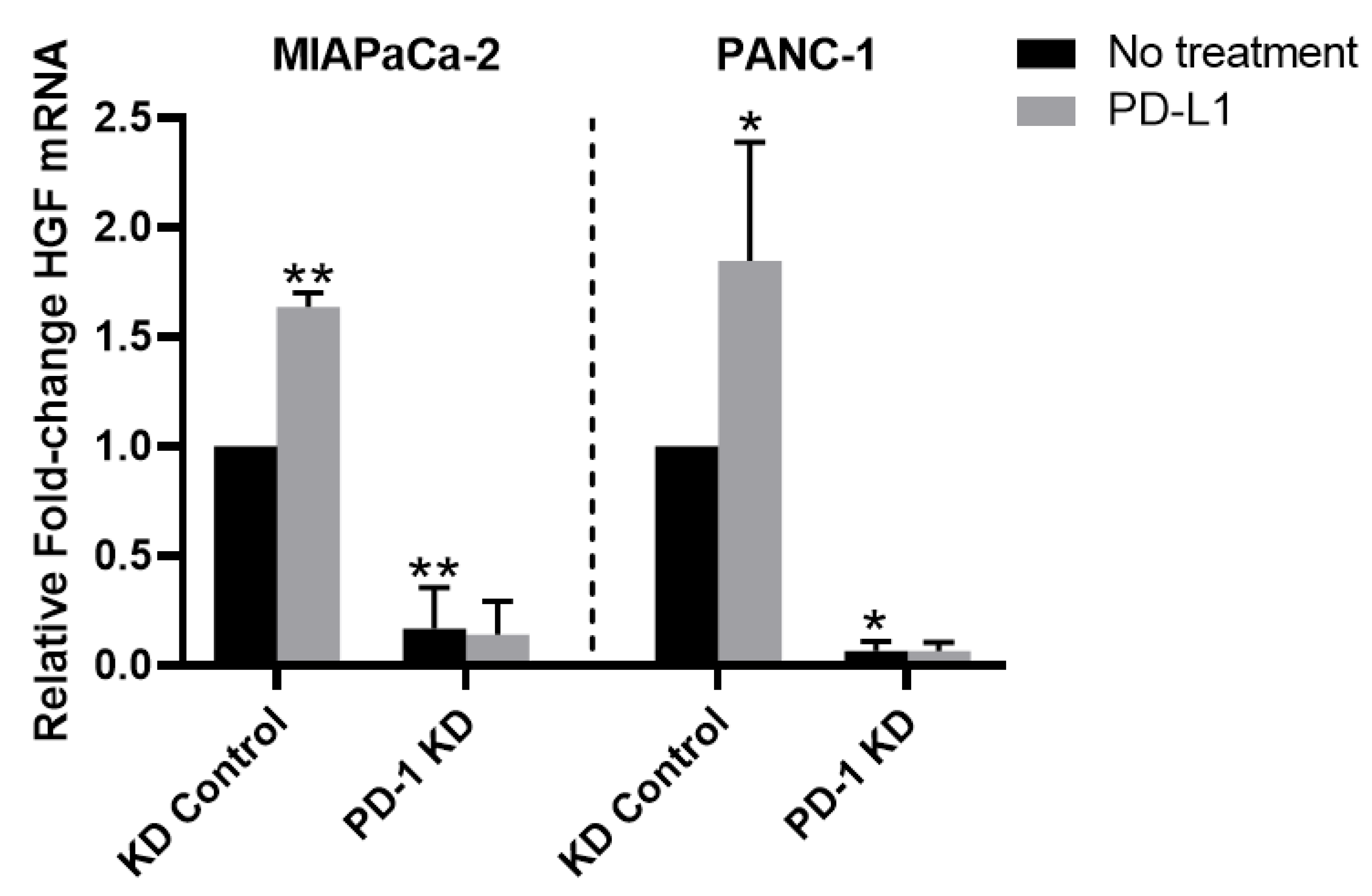
3.6. PD-1/PD-L1 Axis Upregulates HGF mRNA Expression
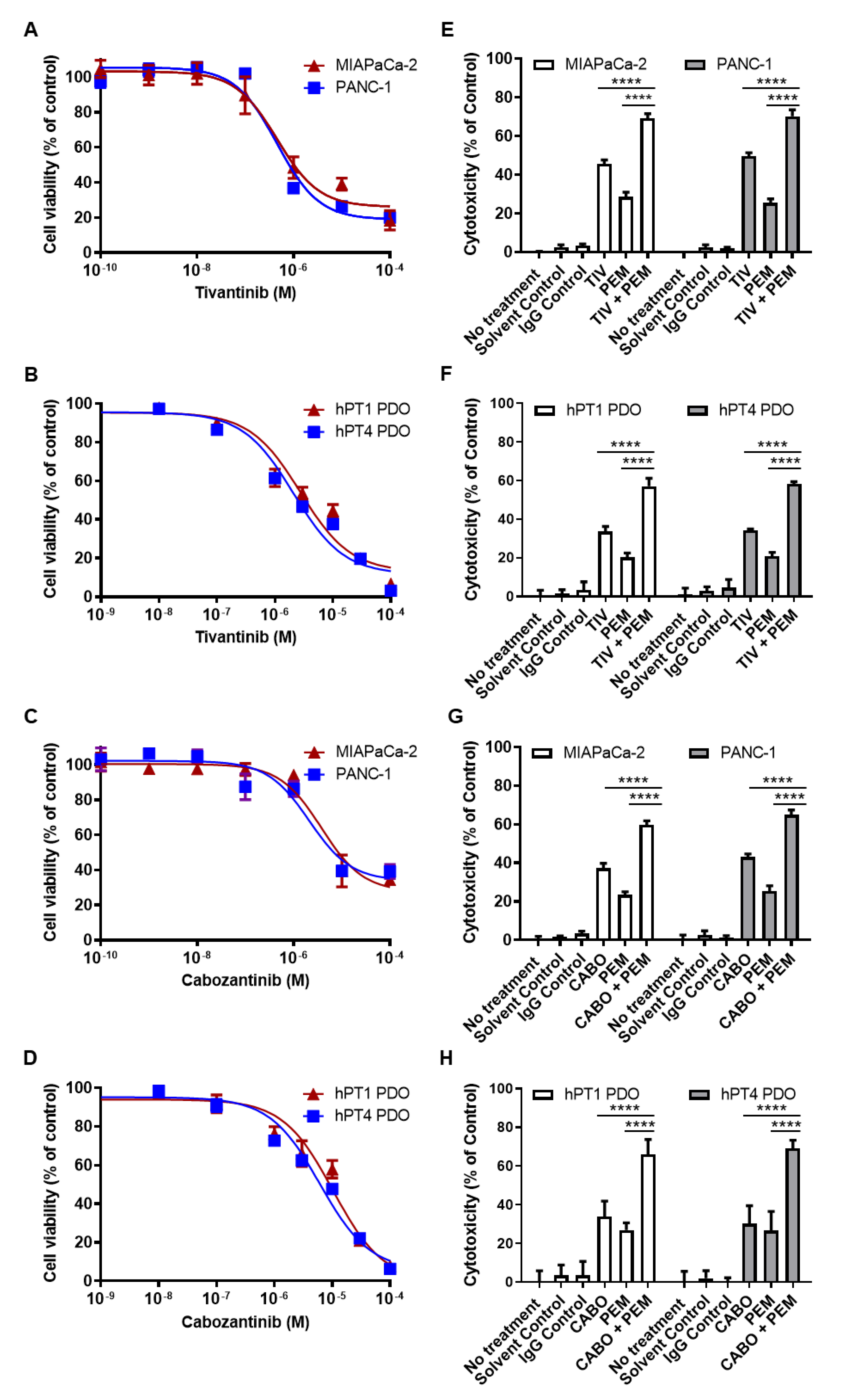
3.7. PD-1 and MET Inhibitors Have Synergistic Cytotoxicity in PDAC Cells and PDOs
3.8. PD-1 and MET Inhibition Effectively Slows Tumor Growth in PDXs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mucciolo, G.; Roux, C.; Scagliotti, A.; Brugiapaglia, S.; Novelli, F.; Cappello, P. The dark side of immunotherapy: Pancreatic cancer. Cancer Drug Resist. 2020, 3, 491–520. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Lin, M.; Moffitt, R.A.; Salazar, M.A.; Park, J.; Vacirca, J.; Huang, C.; Shroyer, K.R.; Choi, M.; Georgakis, G.V.; et al. Direct therapeutic targeting of immune checkpoint PD-1 in pancreatic cancer. Br. J. Cancer 2019, 120, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Eso, Y.; Seno, H. Current status of treatment with immune checkpoint inhibitors for gastrointestinal, hepatobiliary, and pancreatic cancers. Therap. Adv. Gastroenterol. 2020, 13, 1756284820948773. [Google Scholar] [CrossRef] [PubMed]
- Hudson, K.; Cross, N.; Jordan-Mahy, N.; Leyland, R. The Extrinsic and Intrinsic Roles of PD-L1 and Its Receptor PD-1: Implications for Immunotherapy Treatment. Front. Immunol. 2020, 11, 568931. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; O’Reilly, E.M. New Treatment Strategies for Metastatic Pancreatic Ductal Adenocarcinoma. Drugs 2020, 80, 647–669. [Google Scholar] [CrossRef]
- Davern, M.; RM, O.B.; McGrath, J.; Donlon, N.E.; Melo, A.M.; Buckley, C.E.; Sheppard, A.D.; Reynolds, J.V.; Lynam-Lennon, N.; Maher, S.G.; et al. PD-1 blockade enhances chemotherapy toxicity in oesophageal adenocarcinoma. Sci. Rep. 2022, 12, 3259. [Google Scholar] [CrossRef]
- Pu, N.; Gao, S.; Yin, H.; Li, J.-A.; Wu, W.; Fang, Y.; Zhang, L.; Rong, Y.; Xu, X.; Wang, D.; et al. Cell-intrinsic PD-1 promotes proliferation in pancreatic cancer by targeting CYR61/CTGF via the hippo pathway. Cancer Lett. 2019, 460, 42–53. [Google Scholar] [CrossRef]
- Varayathu, H.; Sarathy, V.; Thomas, B.E.; Mufti, S.S.; Naik, R. Combination Strategies to Augment Immune Check Point Inhibitors Efficacy-Implications for Translational Research. Front. Oncol. 2021, 11, 559161. [Google Scholar] [CrossRef]
- Shao, Z.; Pan, H.; Tu, S.; Zhang, J.; Yan, S.; Shao, A. HGF/c-Met Axis: The Advanced Development in Digestive System Cancer. Front. Cell Dev. Biol. 2020, 8, 801. [Google Scholar] [CrossRef]
- Faiella, A.; Riccardi, F.; Carteni, G.; Chiurazzi, M.; Onofrio, L. The Emerging Role of c-Met in Carcinogenesis and Clinical Implications as a Possible Therapeutic Target. J. Oncol. 2022, 2022, 5179182. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, H.S.; Kim, B.J.; Lee, J.; Jang, H.J. Prognostic value of c-Met overexpression in pancreatic adenocarcinoma: A meta-analysis. Oncotarget 2017, 8, 73098–73104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Lin, M.; Rao, M.; Thompson, H.; Hirai, K.; Choi, M.; Georgakis, G.V.; Sasson, A.R.; Bucobo, J.C.; Tzimas, D.; et al. Development of Patient-Derived Gastric Cancer Organoids from Endoscopic Biopsies and Surgical Tissues. Ann. Surg. Oncol. 2018, 25, 2767–2775. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Harper, M.M.; Lin, M.; Qasem, S.A.; Patel, R.A.; Mardini, S.H.; Gabr, M.M.; Cavnar, M.J.; Pandalai, P.K.; Kim, J. Development of a Single-Cell Technique to Increase Yield and Use of Gastrointestinal Cancer Organoids for Personalized Medicine Application. J. Am. Coll. Surg. 2021, 232, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.V.; Nyberg, K.D.; Scott, M.B.; Welsh, A.M.; Nguyen, A.H.; Wu, N.; Hohlbauch, S.V.; Geisse, N.A.; Gibb, E.A.; Robertson, A.G.; et al. Stiffness of pancreatic cancer cells is associated with increased invasive potential. Integr. Biol. 2016, 8, 1232–1245. [Google Scholar] [CrossRef] [Green Version]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res 2017, 23, 3489–3498. [Google Scholar] [CrossRef] [Green Version]
- Harper, M.M.; Lin, M.; Pandalai, P.; Cavnar, M.J.; Gao, M.; Kim, J. Programmed Cell Death-1 Induces MET Activation and Dual Inhibition Synergistically Blocks Pancreatic Cancer Progression. J. Am. Coll. Surg. 2021, 233, S240. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Foster, N.R.; Kim, G.P.; Meyers, J.P.; Smyrk, T.C.; McCullough, A.E.; Ames, M.M.; Jaffe, J.P.; Alberts, S.R. A Phase II Randomized Trial of Panitumumab, Erlotinib, and Gemcitabine Versus Erlotinib and Gemcitabine in Patients with Untreated, Metastatic Pancreatic Adenocarcinoma: North Central Cancer Treatment Group Trial N064B (Alliance). Oncologist 2019, 24, 589-e160. [Google Scholar] [CrossRef] [Green Version]
- Hammel, P.; Huguet, F.; van Laethem, J.L.; Goldstein, D.; Glimelius, B.; Artru, P.; Borbath, I.; Bouche, O.; Shannon, J.; Andre, T.; et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine With or Without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 2016, 315, 1844–1853. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Gagliano, N.; Volpari, T.; Clerici, M.; Pettinari, L.; Barajon, I.; Portinaro, N.; Colombo, G.; Milzani, A.; Dalle-Donne, I.; Martinelli, C. Pancreatic cancer cells retain the epithelial-related phenotype and modify mitotic spindle microtubules after the administration of ukrain in vitro. Anticancer. Drugs 2012, 23, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Gagliano, N.; Celesti, G.; Tacchini, L.; Pluchino, S.; Sforza, C.; Rasile, M.; Valerio, V.; Laghi, L.; Conte, V.; Procacci, P. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma: Characterization in a 3D-cell culture model. World J. Gastroenterol. 2016, 22, 4466–4483. [Google Scholar] [CrossRef] [PubMed]
- Hotz, B.; Arndt, M.; Dullat, S.; Bhargava, S.; Buhr, H.J.; Hotz, H.G. Epithelial to mesenchymal transition: Expression of the regulators snail, slug, and twist in pancreatic cancer. Clin. Cancer Res. 2007, 13, 4769–4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Grünwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592.e18. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef]
- Heinrich, E.L.; Lee, W.; Lu, J.; Lowy, A.M.; Kim, J. Chemokine CXCL12 activates dual CXCR4 and CXCR7-mediated signaling pathways in pancreatic cancer cells. J. Transl. Med. 2012, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yip, M.L.; Shen, X.; Li, H.; Hsin, L.Y.; Labarge, S.; Heinrich, E.L.; Lee, W.; Lu, J.; Vaidehi, N. Identification of anti-malarial compounds as novel antagonists to chemokine receptor CXCR4 in pancreatic cancer cells. PLoS ONE 2012, 7, e31004. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Artinyan, A.; Jackson, D.; Thomas, R.M.; Lowy, A.M.; Kim, J. Chemokine receptor CXCR4 enhances proliferation in pancreatic cancer cells through AKT and ERK dependent pathways. Pancreas 2010, 39, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Heinrich, E.L.; Li, L.; Lu, J.; Choi, A.H.; Levy, R.A.; Wagner, J.E.; Yip, M.L.; Vaidehi, N.; Kim, J. CCR9-mediated signaling through β-catenin and identification of a novel CCR9 antagonist. Mol. Oncol. 2015, 9, 1599–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2020, 468, 72–81. [Google Scholar] [CrossRef]
- Wang, Q.-W.; Sun, L.-H.; Zhang, Y.; Wang, Z.; Zhao, Z.; Wang, Z.-L.; Wang, K.-Y.; Li, G.-Z.; Xu, J.-B.; Ren, C.-Y.; et al. MET overexpression contributes to STAT4-PD-L1 signaling activation associated with tumor-associated, macrophages-mediated immunosuppression in primary glioblastomas. J. Immunother. Cancer 2021, 9, e002451. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Wang-Gillam, A.; Li, C.P.; Bodoky, G.; Dean, A.; Shan, Y.S.; Jameson, G.; Macarulla, T.; Lee, K.H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harper, M.M.; Lin, M.; Qasem, S.A.; Patel, R.A.; Cavnar, M.J.; Pandalai, P.K.; Gao, M.; Kim, J. Endogenous Pancreatic Cancer Cell PD-1 Activates MET and Induces Epithelial-Mesenchymal Transition to Promote Cancer Progression. Cancers 2022, 14, 3051. https://doi.org/10.3390/cancers14133051
Harper MM, Lin M, Qasem SA, Patel RA, Cavnar MJ, Pandalai PK, Gao M, Kim J. Endogenous Pancreatic Cancer Cell PD-1 Activates MET and Induces Epithelial-Mesenchymal Transition to Promote Cancer Progression. Cancers. 2022; 14(13):3051. https://doi.org/10.3390/cancers14133051
Chicago/Turabian StyleHarper, Megan M., Miranda Lin, Shadi A. Qasem, Reema A. Patel, Michael J. Cavnar, Prakash K. Pandalai, Mei Gao, and Joseph Kim. 2022. "Endogenous Pancreatic Cancer Cell PD-1 Activates MET and Induces Epithelial-Mesenchymal Transition to Promote Cancer Progression" Cancers 14, no. 13: 3051. https://doi.org/10.3390/cancers14133051
APA StyleHarper, M. M., Lin, M., Qasem, S. A., Patel, R. A., Cavnar, M. J., Pandalai, P. K., Gao, M., & Kim, J. (2022). Endogenous Pancreatic Cancer Cell PD-1 Activates MET and Induces Epithelial-Mesenchymal Transition to Promote Cancer Progression. Cancers, 14(13), 3051. https://doi.org/10.3390/cancers14133051