
Humanized Ovarian Cancer Patient-Derived Xenografts for Improved Preclinical Evaluation of Immunotherapies

 , , , , ,
, , , , ,  and
and
Abstract
:Simple Summary
Abstract

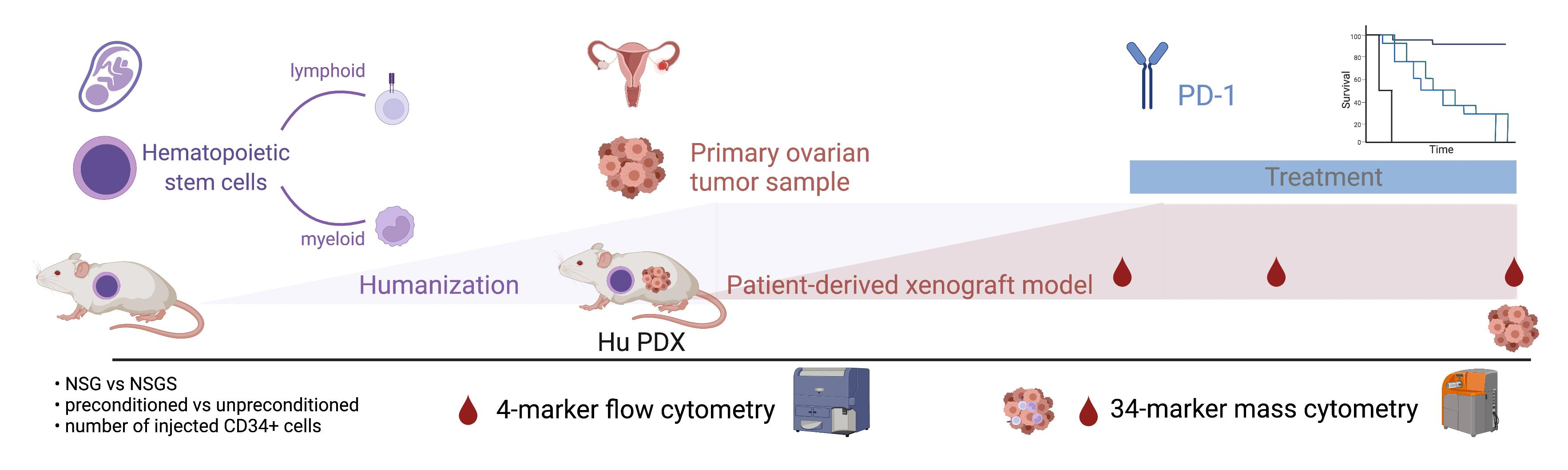
1. Introduction
2. Materials and Methods
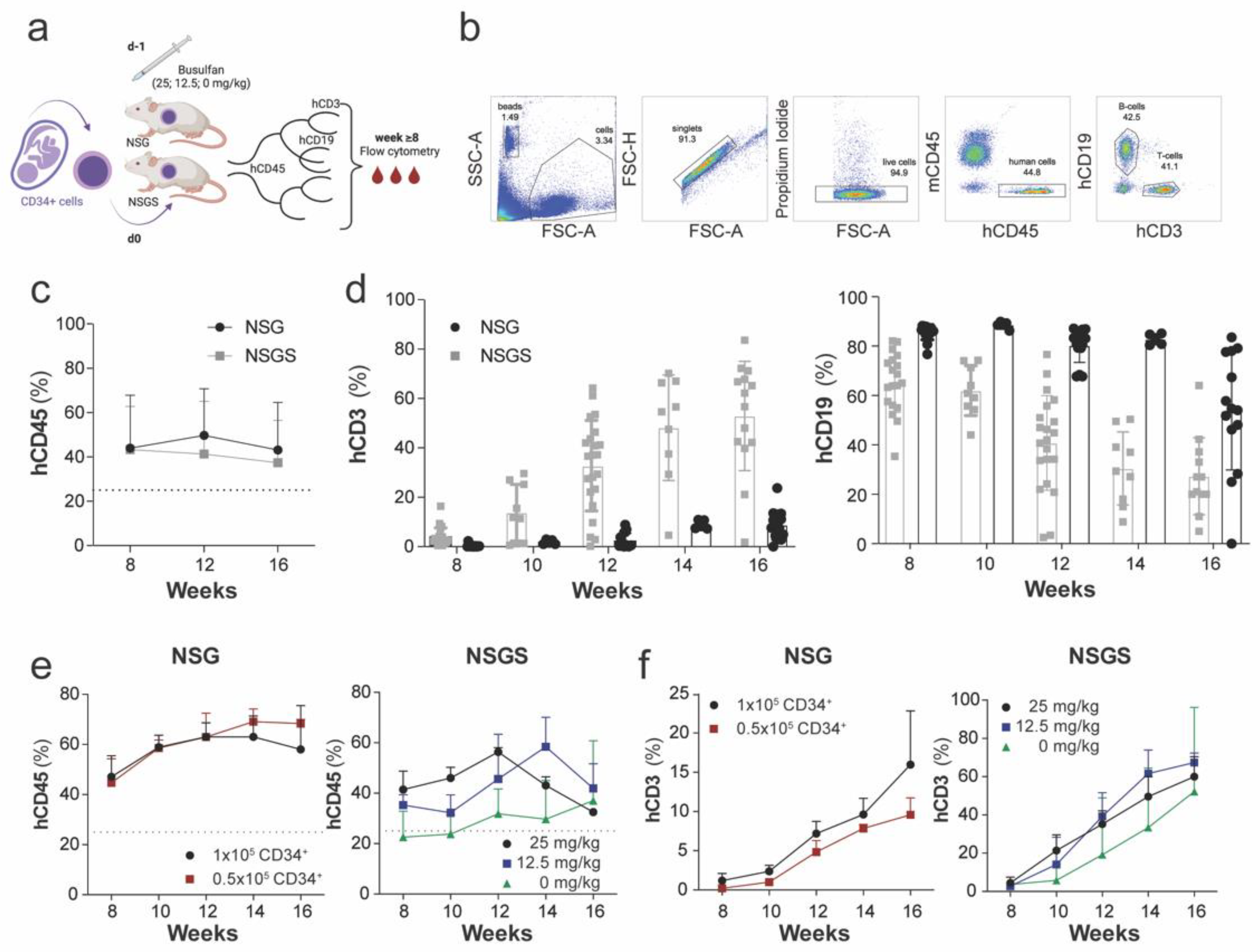
2.1. Generation of Humanized NSG and NSGS Mice
2.2. Generation of Humanized Xenograft Models
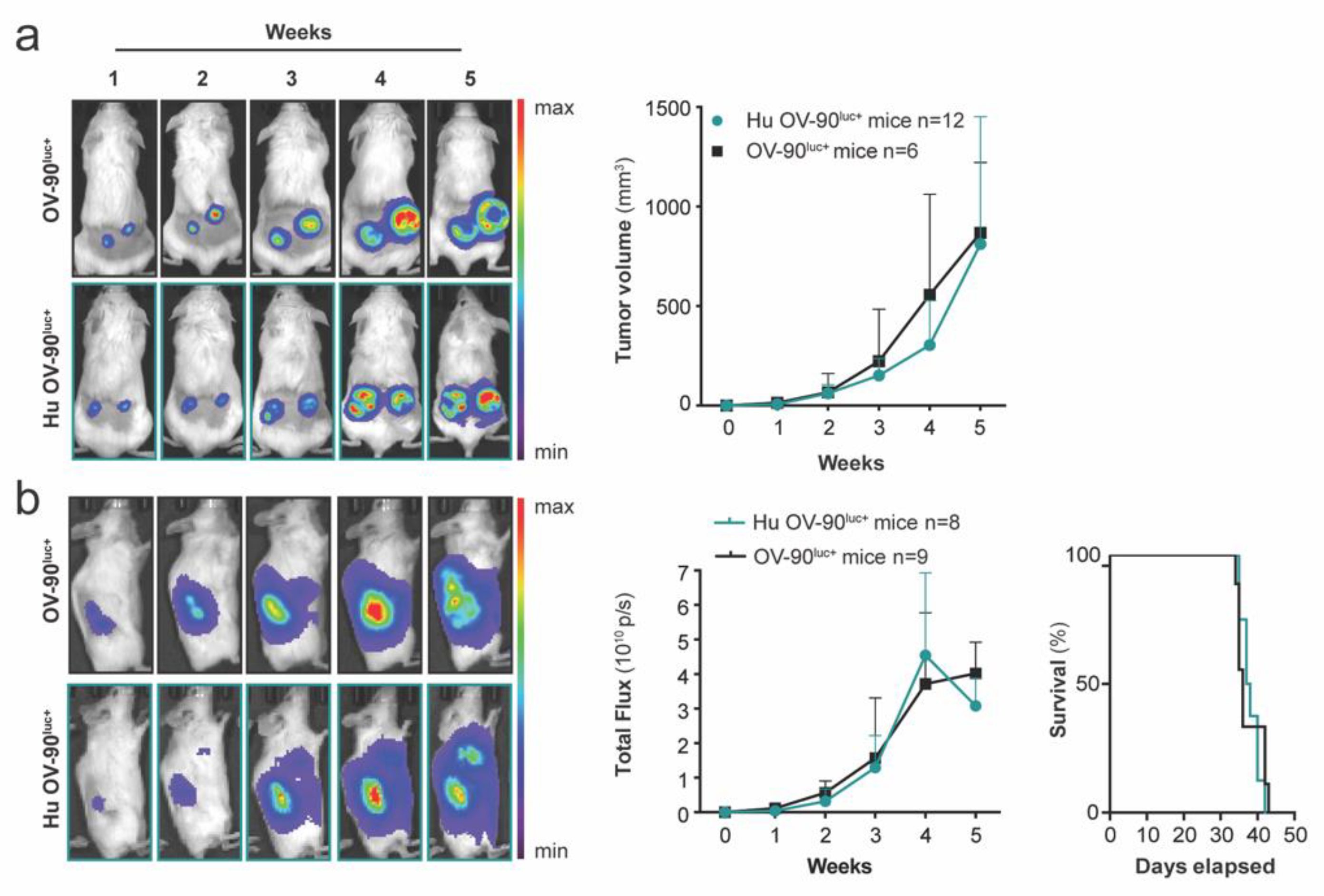
2.3. Evaluation of Tumor Growth
2.4. Treatment
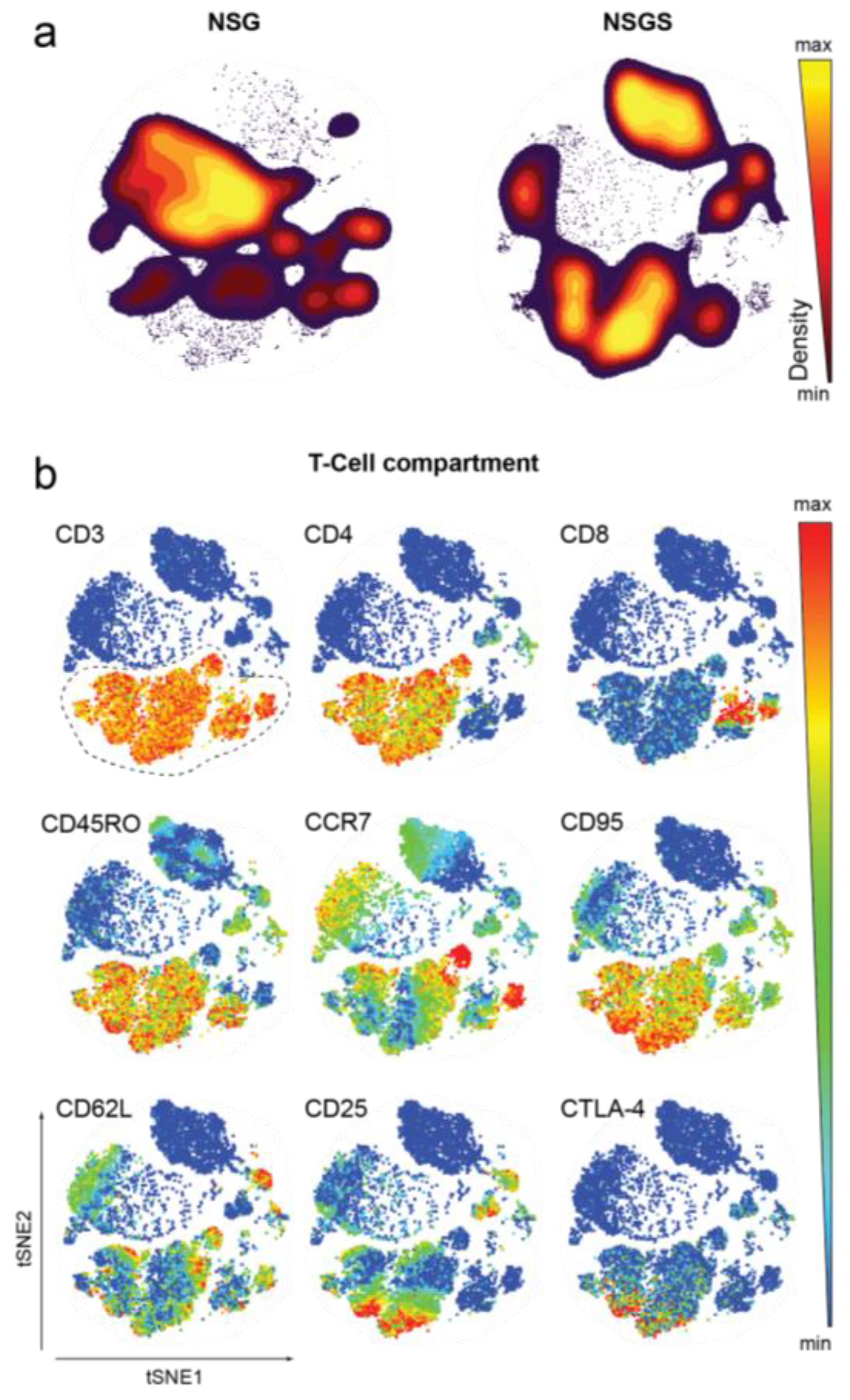
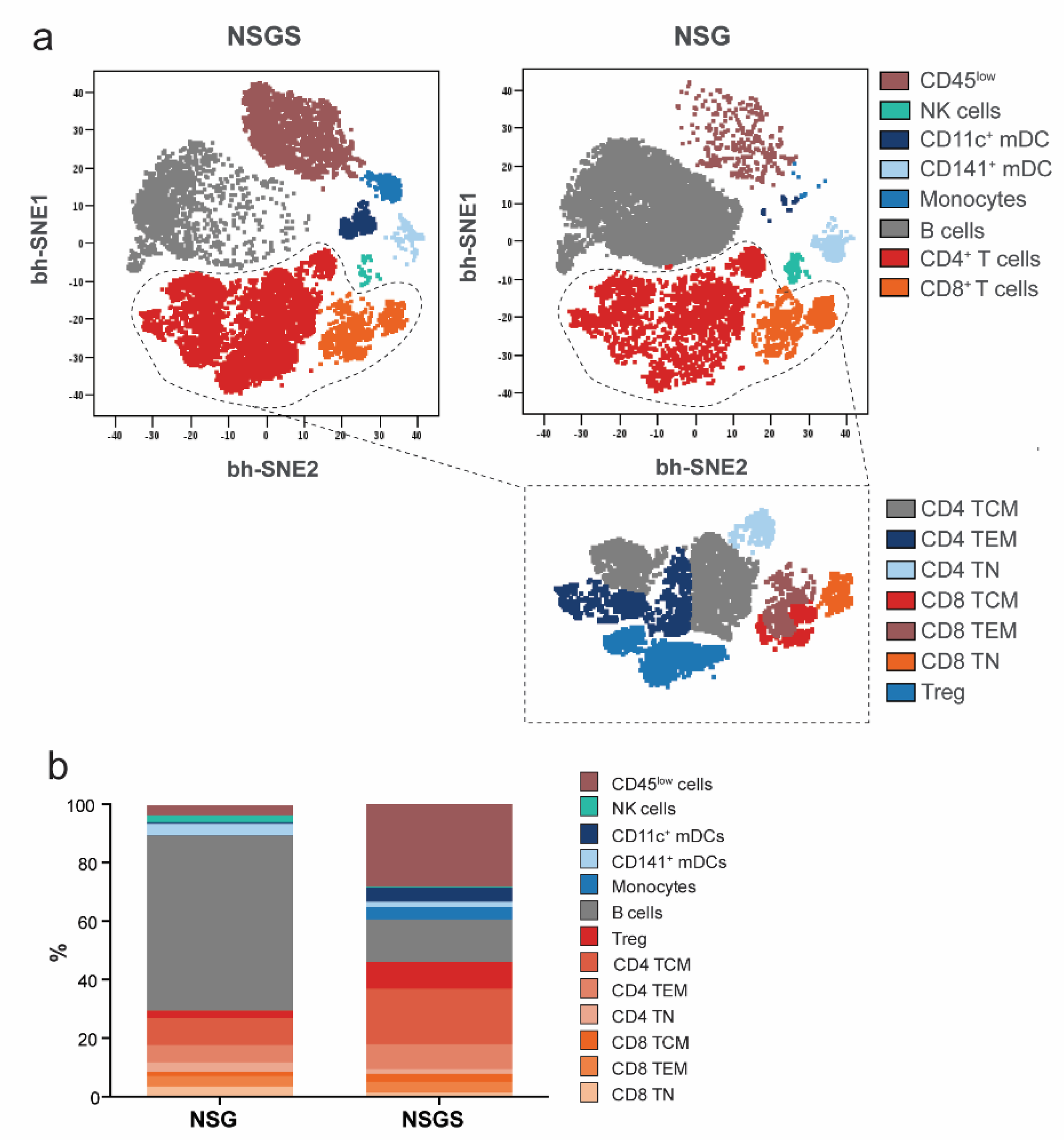
2.5. Evaluation of Immune Cell Subsets
2.6. Flow Cytometry
2.7. Mass Cytometry
2.8. Immunohistochemistry
2.9. Data Analysis
2.10. Statistical Analyses
3. Results
3.1. Parameters Influencing Human Hematopoiesis of Humanized NSG and NSGS Mice
3.2. Characterization of Human Hematopoietic Cells in Peripheral Blood of Hu Mice
3.3. Subcutaneous and Orthotopic Tumor Growth Was Not Impaired in Humanized Mice
3.4. PD-1 Blockade Inhibited Tumor Growth in Hu OV-90 Xenograft and Hu PDX Models
3.5. Characterization of Tumor-Infiltrating Immune Cells after Nivolumab Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial Ovarian Cancer: Evolution of Management in the Era of Precision Medicine. CA Cancer J. Clin. 2019, 393, 1240–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giornelli, G.H. Management of relapsed ovarian cancer: A review. Springerplus 2016, 5, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odunsi, K. Immunotherapy in ovarian cancer. Ann. Oncol. 2017, 28, viii1–viii7. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Liebman, Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [Green Version]
- Duraiswamy, J.; Turrini, R.; Minasyan, A.; Barras, D.; Crespo, I.; Grimm, A.J.; Casado, J.; Genolet, R.; Benedetti, F.; Wicky, A.; et al. Myeloid antigen-presenting cell niches sustain antitumor T cells and license PD-1 blockade via CD28 costimulation. Cancer Cell 2021, 39, 1623.e20–1642.e20. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and antitumor activity of anti–PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef]
- Disis, M.L.; Taylor, M.H.; Kelly, K.; Beck, J.T.; Gordon, M.; Moore, K.M.; Patel, M.R.; Chaves, J.; Park, H.; Mita, A.C.; et al. Efficacy and Safety of Avelumab for Patients With Recurrent or Refractory Ovarian Cancer: Phase 1b Results From the JAVELIN Solid Tumor Trial. JAMA Oncol. 2019, 5, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Results from the phase II KEYNOTE-100 study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, G.M.; Galpin, K.J.; McCloskey, C.W.; Vanderhyden, B.C. The Tumor Microenvironment of Epithelial Ovarian Cancer and Its Influence on Response to Immunotherapy. Cancers 2018, 10, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz-scid IL2R null Mice Engrafted with Mobilized Human Hemopoietic Stem Cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.N.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized mouse models of clinical disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 187–215. [Google Scholar] [CrossRef] [Green Version]
- Wunderlich, M.; Chou, F.S.; Link, K.A.; Mizukawa, B.; Perry, R.L.; Carroll, M.; Mulloy, J.C. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia 2010, 24, 1785–1788. [Google Scholar] [CrossRef] [Green Version]
- Arranz, L. The Hematology of Tomorrow Is Here—Preclinical Models Are Not: Cell Therapy for Hematological Malignancies. Cancers 2022, 14, 580. [Google Scholar] [CrossRef]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef]
- Billerbeck, E.; Barry, W.T.; Mu, K.; Dorner, M.; Rice, C.M.; Plos, A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rgamma(null) humanized mice. Blood 2011, 117, 3076–3086. [Google Scholar] [CrossRef] [Green Version]
- Wunderlich, M.; Stockman, C.; Devarajan, M.; Ravishankar, N.; Sexton, C.; Kumar, A.R.; Mizukawa, B.; Mulloy, J.C. A xenograft model of macrophage activation syndrome amenable to anti-CD33 and anti-IL-6R treatment. JCI Insight 2016, 1, e88181. [Google Scholar] [CrossRef] [Green Version]
- Audigé, A.; Rochat, M.A.; Li, D.; Ivic, S.; Fahrny, A.; Muller, C.K.; Gers-Huber, G.; Myburgh, R.; Bredl, S.; Schlaepfer, E.; et al. Long-term leukocyte reconstitution in NSG mice transplanted with human cord blood hematopoietic stem and progenitor cells. BMC Immunol. 2017, 18, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, I.A.; Muench, M.O. Comparison of Human Hematopoietic Reconstitution in Different Strains of Immunodeficient Mice. Stem. Cells Dev. 2017, 26, 102–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strowig, T.; Rongvaux, A.; Rathinam, C.; Takizawa, H.; Borsotti, C.; Philbrick, W.; Eynon, E.E.; Manz, M.G.; Flavell, R.A. Transgenic expression of human signal regulatory protein alpha in Rag2-/-gamma(c)-/- mice improves engraftment of human hematopoietic cells in humanized mice. Proc. Natl. Acad. Sci. USA 2011, 108, 13218–13223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoda, Y.; Virshup, I.; Rojas, I.L.; Haigh, O.; Wong, Y.; Miles, J.J.; Wells, C.A.; Radford, K.J. Human CD141(+) Dendritic Cell and CD1c(+) Dendritic Cell Undergo Concordant Early Genetic Programming after Activation in Humanized Mice In Vivo. Front. Immunol. 2017, 8, 1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankert, B.R.; Balu-Iyer, S.V.; Odunsi, K.; Shultz, L.D.; Kelleher, R.J., Jr.; Barnas, J.L.; Simpson-Abelson, M.; Parsons, R.; Yokota, S.J. Humanized mouse model of ovarian cancer recapitulates patient solid tumor progression, ascites formation, and metastasis. PLoS ONE 2011, 6, e24420. [Google Scholar] [CrossRef] [Green Version]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.-K.; Peterson, E.; Sun, J.; Goudie, C.; Drapkin, R.I.; Liu, J.F.; Matulonis, U.; Zhu, Q.; Marasco, W.A. Anti-CCR4 monoclonal antibody enhances antitumor immunity by modulating tumor-infiltrating Tregs in an ovarian cancer xenograft humanized mouse model. Oncoimmunology 2016, 5, e1090075. [Google Scholar] [CrossRef] [Green Version]
- Odunsi, A.; McGray, A.R.; Miliotto, A.; Zhang, Y.; Wang, J.; Abiola, A.; Eppolito, C.; Huang, R.-Y. Fidelity of human ovarian cancer patient-derived xenografts in a partially humanized mouse model for preclinical testing of immunotherapies. J. Immunother. Cancer 2020, 8, e001237. [Google Scholar] [CrossRef]
- Kleinmanns, K.; Bischof, K.; Anandan, S.; Popa, M.; Akslen, L.A.; Fosse, V.; Karlsen, I.; Gjertsen, B.T.; Bjorge, L.; McCormack, E. CD24-targeted fluorescence imaging in patient-derived xenograft models of high-grade serous ovarian carcinoma. EBioMedicine 2020, 56, 102782. [Google Scholar] [CrossRef]
- Zunder, R.E.; Finck, R.; Behbehani, G.K.; El-ad, D.A.; Krishnaswamy, S.; Gonzalez, V.D.; Lorang, C.G.; Bjornson, Z.; Spitzer, M.H.; Bodenmiller, B. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc. 2015, 10, 316. [Google Scholar] [CrossRef]
- Bendall, S.C.; Simonds, E.F.; Qiu, P.; Amir, E.A.D.; Krutzik, P.O.; Finck, R.; Bruggner, R.V.; Melamed, R.; Trejo, A.; Ornatsky, O.I.; et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011, 332, 687–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir el, A.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, P.; Simonds, E.F.; Bendall, S.C.; Gibbs, K.D., Jr.; Bruggner, R.V.; Linderman, M.D.; Sachs, K.; Nolan, G.P.; Plevritis, S.K. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol. 2011, 29, 886–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.R.; Thakkar, K.N.; Qian, J.; Kariolis, M.S.; Huang, W.; Nandagopal, S.; Yang, T.T.C.; Diep, A.N.; Cherf, G.M.; Xu, Y. Neutralization of PD-L2 is essential for overcoming immune checkpoint blockade resistance in ovarian cancer. Clin. Cancer Res. 2021, 27, 4435–4448. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The next decade of immune checkpoint therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef]
- Shultz, L.D.; Saito, Y.; Najima, Y.; Tanaka, S.; Ochi, T.; Tomizawa, M.; Doi, T.; Sone, A.; Suzuki, N.; Fujiwara, H.; et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc. Natl. Acad. Sci. USA 2010, 107, 13022–13027. [Google Scholar] [CrossRef] [Green Version]
- Melkus, M.W.; Estes, J.D.; Padgett-Thomas, A.; Gatlin, J.; Denton, P.W.; Othieno, F.A.; Wege, A.K.; Haase, A.T.; Garcia, J.V. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat. Med. 2006, 12, 1316. [Google Scholar] [CrossRef]
- Saito, Y.; Ellegast, J.M.; Rafiei, A.; Song, Y.; Kull, D.; Heikenwalder, M.; Rongvaux, A.; Halene, S.; Flavell, R.A.; Manz, M.G. Peripheral blood CD34+ cells efficiently engraft human cytokine knock-in mice. Blood 2016, 128, 1829–1833. [Google Scholar] [CrossRef] [Green Version]
- Herndler-Brandstetter, D.; Shan, L.; Yao, Y.; Stecher, C.; Plajer, V.; Lietzenmayer, M.; Strowig, T.; De Zoete, M.R.; Palm, N.W.; Chen, J.; et al. Humanized mouse model supports development, function, and tissue residency of human natural killer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E9626–E9634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, F.E.; Cashman, J.D.; Hogge, D.E.; Humphries, R.K.; Eaves, C.J. NOD/SCID mice engineered to express human IL-3, GM-CSF and Steel factor constitutively mobilize engrafted human progenitors and compromise human stem cell regeneration. Leukemia 2004, 18, 341–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmig, S.; Kronstein-Wiedemann, R.; Fohgrub, J.; Kronstein, N.; Nevmerzhitskaya, A.; Bornhäuser, M.; Gassmann, M.; Platz, A.; Ordemann, R.; Tonn, T. Improved human erythropoiesis and platelet formation in humanized NSGW41 mice. Stem Cell Rep. 2016, 7, 591–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rongvaux, A.; Willinger, T.; Takizawa, H.; Rathinam, C.; Auerbach, W.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Eynon, E.E.; Stevens, S. Human thrombopoietin knockin mice efficiently support human hematopoiesis in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 2378–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Shultz, L.D.; Ishikawa, F. Understanding normal and malignant human hematopoiesis using next-generation humanized mice. Trends Immunol. 2020, 41, 706–720. [Google Scholar] [CrossRef]
- Wang, M.; Yao, L.C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.X.; Shi, W.; Ma, A.H.; de Vere White, R.W.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef] [Green Version]
- Capasso, A.; Lang, J.; Pitts, T.; Jordan, K.; Lieu, C.; Davis, S.; Diamond, J.; Kopetz, S.; Barbee, J.; Peterson, J. Characterization of immune responses to anti-PD-1 mono and combination immunotherapy in hematopoietic humanized mice implanted with tumor xenografts. J. Immunother. Cancer 2019, 7, 37. [Google Scholar] [CrossRef]
- Tary-Lehmann, M.; Saxon, A.; Lehmann, P.V. The human immune system in hu-PBL-SCID mice. Immunol. Today 1995, 16, 529–533. [Google Scholar] [CrossRef]
- Ali, N.; Flutter, B.; Rodriguez, R.S.; Sharif-Paghaleh, E.; Barber, L.D.; Lombardi, G.; Nestle, F.O. Xenogeneic graft-versus-host-disease in NOD-scid IL-2Rγnull mice display a T-effector memory phenotype. PLoS ONE 2012, 7, e44219. [Google Scholar]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.A.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120.e17–1133.e17. [Google Scholar] [CrossRef] [Green Version]
- Normann, M.C.; Türzer, M.; Diep, L.M.; Oldenburg, J.; Gajdzik, B.; Solheim, O.; Rud, E. Early experiences with PD-1 inhibitor treatment of platinum resistant epithelial ovarian cancer. J. Gynecol. Oncol. 2019, 30, e56. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Cannistra, S.A. Immune Checkpoint Inhibitors in Ovarian Cancer: Can We Bridge the Gap Between IMagynation and Reality? J. Clin. Oncol. 2021, 39, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, e143–e152. [Google Scholar] [CrossRef] [Green Version]
- Ning, F.; Cole, C.B.; Annunziata, C.M. Driving immune responses in the ovarian tumor microenvironment. Front. Oncol. 2021, 10, 604084. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhang, J.; Li, D.; Mao, Y.; Mo, F.; Du, W.; Ma, X. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2017, 147, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Drakes, M.L.; Stiff, P.J. Regulation of ovarian cancer prognosis by immune cells in the tumor microenvironment. Cancers 2018, 10, 302. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, K.; Komohara, Y.; Takaishi, K.; Katabuchi, H.; Takeya, M. Detection of M2 macrophages and colony-stimulating factor 1 expression in serous and mucinous ovarian epithelial tumors. Pathol. Int. 2009, 59, 300–305. [Google Scholar] [CrossRef]
- Helland, O.; Popa, M.; Bischof, K.; Gjertsen, B.T.; McCormack, E.; Bjorge, L. The HDACi Panobinostat Shows Growth Inhibition Both In Vitro and in a Bioluminescent Orthotopic Surgical Xenograft Model of Ovarian Cancer. PLoS ONE 2016, 11, e0158208. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Waggoner, S.E.; Vidal, G.A.; Mita, M.M.; Fleming, G.F.; Holloway, R.W.; van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib+ pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. J. Clin. Oncol. 2018, 36, 106. [Google Scholar] [CrossRef]
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, A.; Hernandez, D.C.; Schallmoser, K.; Majeti, R. Generation and use of a humanized bone-marrow-ossicle niche for hematopoietic xenotransplantation into mice. Nat. Protoc. 2017, 12, 2169–2188. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NSGS | NSG | NSGS Hu | NSG Hu | ||||
|---|---|---|---|---|---|---|---|
| Week | Busulfan Concentration (mg/kg) | ||||||
| 0 | 12.5 | 25 | 25 | ||||
| RBC (M/μL) | 8.0 ± 0.32 | 10 | 6.28 ± 0.12 | 4.75 ± 1.78 | 4.70 ± 0.71 | 8.12 ± 0.30 | |
| 8.8 ± 0.54 | 14 | 4.74 ± 0.81 | 3.99 ± 0.53 | 3.84 ± 0.76 | 7.22 ± 0.34 | ||
| 18 | 3.04 ± 1.98 | 2.31 ± 0.38 | 3.45 ± 0.06 | 5.92 ± 0.99 | |||
| HGB (g/dl) | 13.48 ± 0.51 | 10 | 12.1 ± 0.4 | 9.30 ± 3.38 | 9.67 ± 1.12 | 13.50 ± 0.5 | |
| 13.96 ± 0.94 | 14 | 9.83 ± 1.55 | 8.50 ± 0.82 | 8.3 ± 1.01 | 12.72 ± 0.63 | ||
| 18 | 6.73 ± 4.05 | 5.40 ± 0.99 | 7.75 ± 0.21 | 10.74 ± 1.85 | |||
| Platelets (K/μL) | 1007.4 ± 69.2 | 10 | 635.3 ± 41.2 | 406.3 ± 174.2 | 296 ± 103.5 | 508.3 ± 52.8 | |
| 1093.4 ± 94.6 | 14 | 550.0 ± 225.4 | 280.0 ±1 33.4 | 315.0 ± 109.3 | 619.0 ± 104.7 | ||
| 18 | 228.0 ± 143.4 | 174.0 ± 144.3 | 222.0 ± 18.4 | 544.2 ± 114.0 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleinmanns, K.; Gullaksen, S.-E.; Bredholt, G.; Davidson, B.; Torkildsen, C.F.; Grindheim, S.; Bjørge, L.; McCormack, E. Humanized Ovarian Cancer Patient-Derived Xenografts for Improved Preclinical Evaluation of Immunotherapies. Cancers 2022, 14, 3092. https://doi.org/10.3390/cancers14133092
Kleinmanns K, Gullaksen S-E, Bredholt G, Davidson B, Torkildsen CF, Grindheim S, Bjørge L, McCormack E. Humanized Ovarian Cancer Patient-Derived Xenografts for Improved Preclinical Evaluation of Immunotherapies. Cancers. 2022; 14(13):3092. https://doi.org/10.3390/cancers14133092
Chicago/Turabian StyleKleinmanns, Katrin, Stein-Erik Gullaksen, Geir Bredholt, Ben Davidson, Cecilie Fredvik Torkildsen, Sindre Grindheim, Line Bjørge, and Emmet McCormack. 2022. "Humanized Ovarian Cancer Patient-Derived Xenografts for Improved Preclinical Evaluation of Immunotherapies" Cancers 14, no. 13: 3092. https://doi.org/10.3390/cancers14133092
APA StyleKleinmanns, K., Gullaksen, S.-E., Bredholt, G., Davidson, B., Torkildsen, C. F., Grindheim, S., Bjørge, L., & McCormack, E. (2022). Humanized Ovarian Cancer Patient-Derived Xenografts for Improved Preclinical Evaluation of Immunotherapies. Cancers, 14(13), 3092. https://doi.org/10.3390/cancers14133092