
Intronic Polyadenylation in Acquired Cancer Drug Resistance Circumvented by Utilizing CRISPR/Cas9 with Homology-Directed Repair: The Tale of Human DNA Topoisomerase IIα

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
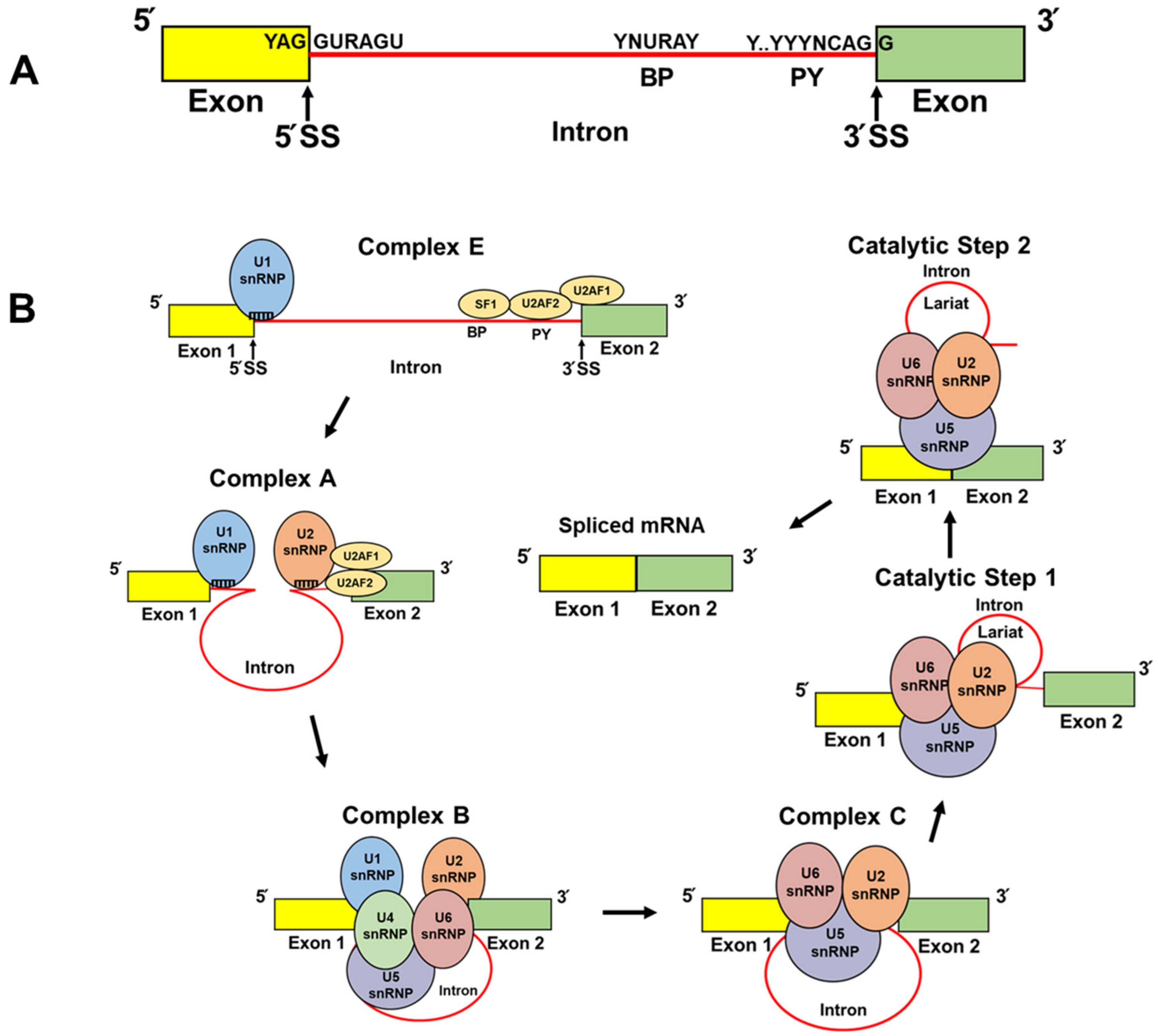
2. The Spliceosome
3. Alternative Splicing
4. Cleavage and Polyadenylation Factors (CPAFs)
5. Alternative Polyadenylation
6. Intronic Polyadenylation (IPA)
7. The Tale (Tail) of TOP2α IPA in Acquired Chemoresistance: Part 1
8. The Tale (Tail) of TOP2α IPA in Acquired Chemoresistance: Part 2
9. TOP2α/90 IPA in K/VP.5 Cells: Using CRISPR/Cas9/HDR to Circumvent Drug Resistance
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, B.H.; Osheroff, N.; Berger, J.M. Structure of a topoisomerase II-DNA-nucleotide complex reveals a new control mechanism for ATPase activity. Nat. Struct. Mol. Biol. 2012, 19, 1147–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Chan, N.L.; Hsieh, T.S. New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem. 2013, 82, 139–170. [Google Scholar] [CrossRef]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Economides, M.P.; McCue, D.; Borthakur, G.; Pemmaraju, N. Topoisomerase II inhibitors in AML: Past, present, and future. Expert Opin. Pharmacother. 2019, 20, 1637–1644. [Google Scholar] [CrossRef]
- Edwardson, D.W.; Narendrula, R.; Chewchuk, S.; Mispel-Beyer, K.; Mapletoft, J.P.J.; Parissenti, A.M. Role of drug metabolism in the cytotoxicity and clinical efficacy of anthracyclines. Curr. Drug Metab. 2015, 16, 412–426. [Google Scholar] [CrossRef] [Green Version]
- Shanbhag, S.; Ambinder, R.F. Hodgkin lymphoma: A review and update on recent progress. CA Cancer J. Clin. 2018, 68, 116–132. [Google Scholar] [CrossRef]
- Pommier, Y.; Marchand, C. Interfacial inhibitors: Targeting macromolecular complexes. Nat. Rev. Drug Discov. 2011, 11, 25–36. [Google Scholar] [CrossRef]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Burgess, D.J.; Doles, J.; Zender, L.; Xue, W.; Ma, B.; McCombie, W.R.; Hannon, G.J.; Lowe, S.W.; Hemann, M.T. Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc. Nat. Acad. Sci. USA 2008, 105, 9053–9058. [Google Scholar] [CrossRef] [Green Version]
- Pilati, P.; Nitti, D.; Mocellin, S. Cancer resistance to type II topoisomerase inhibitors. Curr. Med. Chem. 2012, 19, 3900–3906. [Google Scholar] [CrossRef]
- Ganapathi, R.N.; Ganapathi, M.K. Mechanisms regulating resistance to inhibitors of topoisomerase II. Front. Pharmacol. 2013, 4, 89. [Google Scholar] [CrossRef] [Green Version]
- Capelôa, T.; Benyahia, Z.; Zampieri, L.X.; Blackman, M.C.N.M.; Sonveaux, P. Metabolic and non-metabolic pathways that control cancer resistance to anthracyclines. Semin. Cell Dev. Biol. 2020, 98, 181–191. [Google Scholar] [CrossRef]
- Elton, T.S.; Ozer, H.G.; Yalowich, J.C. Effects of DNA topoisomerase IIα splice variants on acquired drug resistance. Cancer Drug Resist. 2020, 3, 161–170. [Google Scholar] [CrossRef]
- Zahreddine, H.; Borden, K.L. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef] [Green Version]
- Harker, W.G.; Slade, D.L.; Parr, R.L.; Holguin, M.H. Selective use of an alternative stop codon and polyadenylation signal within intron sequences leads to a truncated topoisomerase II alpha messenger RNA and protein in human HL-60 leukemia cells selected for resistance to mitoxantrone. Cancer Res. 1995, 55, 4962–4971. [Google Scholar]
- Mo, Y.Y.; Beck, W.T. Heterogeneous expression of DNA topoisomerase II alpha isoforms in tumor cell lines. Oncol. Res. 1997, 9, 193–204. [Google Scholar]
- Kanagasabai, R.; Serdar, L.; Karmahapatra, S.; Kientz, C.A.; Ellis, J.; Ritke, M.K.; Elton, T.S.; Yalowich, J.C. Alternative RNA processing of topoisomerase II alpha in etoposide-resistant human leukemia K562 cells: Intron retention results in a novel c-terminal truncated 90-kDa isoform. J. Pharmacol. Exp. Ther. 2017, 360, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Kanagasabai, R.; Karmahapatra, S.; Kientz, C.A.; Yu, Y.; Hernandez, V.A.; Kania, E.E.; Yalowich, J.C.; Elton, T.S. The novel C-terminal truncated 90-kDa isoform of topoisomerase II alpha (TOP2α/90) is a determinant of etoposide resistance in K562 leukemia cells via heterodimerization with the TOP2α/170 isoform. Mol. Pharmacol. 2018, 93, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, V.A.; Carvajal-Moreno, J.; Papa, J.L.; Shkolnikov, N.; Li, J.; Ozer, H.G.; Yalowich, J.C.; Elton, T.S. CRISPR/Cas9 Genome Editing of the Human Topoisomerase II α Intron 19 5′ Splice Site Circumvents Etoposide Resistance in Human Leukemia K562 Cells. Mol. Pharmacol. 2021, 99, 226–241. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E.A. Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Potter, J.; Kumar, S.; Ravinder, N.; Chesnut, J.D. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J. Biotechnol. 2017, 241, 136–146. [Google Scholar] [CrossRef]
- Cowling, V.H. Regulation of mRNA cap methylation. Biochem. J. 2009, 425, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.A. Split genes and RNA splicing. Cell 1994, 77, 805–815. [Google Scholar] [CrossRef]
- Sharp, P.A. The discovery of split genes and RNA splicing. Trends. Biochem. Sci. 2005, 30, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Manley, J.L. Alternative cleavage and polyadenylation: The long and short of it. Trends Biochem. Sci. 2013, 38, 312–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Manley, J.L. The end of the message: Multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015, 29, 889–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef]
- Mayr, C. Regulation by 3′-untranslated regions. Annu. Rev. Genet. 2017, 51, 171–194. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative pre-mRNA splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [Green Version]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and mechanisms of alternative splicing in cancer-implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Taggart, A.J.; DeSimone, A.M.; Shih, J.S.; Filloux, M.E.; Fairbrother, W.G. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat. Struct. Mol. Biol. 2012, 19, 719–721. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Barbosa-Morais, N.L.; Irimia, M.; Pan, Q.; Xiong, H.Y.; Gueroussov, S.; Lee, L.J.; Slobodeniuc, V.; Kutter, C.; Watt, S.; Colak, R.; et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012, 338, 1587–1593. [Google Scholar] [CrossRef] [Green Version]
- Monteuuis, G.; Wong, J.J.L.; Bailey, C.G.; Schmitz, U.; Rasko, J.E.J. The changing paradigm of intron retention: Regulation, ramifications and recipes. Nucleic Acids Res. 2019, 47, 11497–11513. [Google Scholar] [CrossRef]
- Boutz, P.L.; Bhutkar, A.; Sharp, P.A. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 2015, 29, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Maquat, L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef]
- Fiszbein, A.; Kornblihtt, A.R. Alternative splicing switches: Important players in cell differentiation. Bioessays 2017, 39, 1600157. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- David, C.J.; Manley, J.L. Alternative pre- mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [Green Version]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.A.; Krainer, A.R.; Abdel-Wahab, O. SnapShot: Splicing alterations in cancer. Cell 2020, 180, 208. [Google Scholar] [CrossRef]
- Hu, J.; Lutz, C.S.; Wilusz, J.; Tian, B. Bioinformatic identification of candidate cis-regulatory elements involved in human mRNA polyadenylation. RNA 2005, 11, 1485–1493. [Google Scholar] [CrossRef] [Green Version]
- Ruegsegger, U.; Blank, D.; Keller, W. Human pre-mRNA cleavage factor Im is related to spliceosomal SR proteins and can be reconstituted in vitro from recombinant subunits. Mol. Cell 1998, 1, 243–253. [Google Scholar] [CrossRef]
- Chan, S.; Huppertz, I.; Yao, C.; Weng, L.; Moresco, J.; Yates, J.R.; Ule, J.; Manley, J.; Shi, Y. CPSF30 and Wdr33 directly bind to AAUAAA in mammalian mRNA 3′ processing. Genes Dev. 2014, 28, 2370–2380. [Google Scholar] [CrossRef] [Green Version]
- Schonemann, L.; Kuhn, U.; Martin, G.; Schafer, P.; Gruber, A.R.; Keller, W.; Zavolan, M.; Wahle, E. Reconstitution of CPSF active in polyadenylation: Recognition of the polyadenylation signal by WDR33. Genes Dev. 2014, 28, 2381–2393. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, I.; Martin, G.; Friedlein, A.; Langen, H.; Keller, W. Human Fip1 is a subunit of CPSF that binds to U-rich RNA elements and stimulates poly (A) polymerase. EMBO J. 2004, 23, 616–626. [Google Scholar] [CrossRef] [Green Version]
- Mandel, C.R.; Kaneko, S.; Zhang, H.; Gebauer, D.; Vethantham, V.; Manley, J.L.; Tong, L. Polyadenylation factor CPSF-73 is the pre-mRNA 3′-end-processing endonuclease. Nature 2006, 444, 953–956. [Google Scholar] [CrossRef]
- Schäfer, P.; Tüting, C.; Schönemann, L.; Kühn, U.; Treiber, T.; Treiber, N.; Ihling, C.; Graber, A.; Keller, W.; Meister, G.; et al. Reconstitution of mammalian cleavage factor II involved in 3′ processing of mRNA precursors. RNA 2018, 24, 1721–1737. [Google Scholar] [CrossRef] [Green Version]
- Derti, A.; Garrett-Engele, P.; Macisaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.; Rohl, C.A.; Johnson, J.M.; Babak, T. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012, 22, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Hoque, M.; Ji, Z.; Zheng, D.; Luo, W.; Li, W.; You, B.; Park, J.Y.; Yehia, G.; Tian, B. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods 2013, 10, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Lianoglou, S.; Garg, V.; Yang, J.L.; Leslie, C.S.; Mayr, C. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 2013, 27, 2380–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamieniarz-Gdula, K.; Proudfoot, N.J. Transcriptional Control by Premature Termination: A Forgotten Mechanism. Trends Genet. 2019, 35, 553–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3′untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef] [Green Version]
- Weill, L.; Belloc, E.; Bava, F.-A.; Méndez, R. Translational control by changes in poly (A) tail length: Recycling mRNAs. Nat. Struct. Mol. Biol. 2012, 19, 577–585. [Google Scholar] [CrossRef]
- Tian, B.; Pan, Z.; Lee, J.Y. Widespread mRNA polyadenylation events in introns indicate dynamic interplay between polyadenylation and splicing. Genome Res. 2007, 17, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; You, B.; Hoque, M.; Zheng, D.; Luo, W.; Ji, Z.; Park, J.Y.; Gunderson, S.I.; Kalsotra, A.; Manley, J.L.; et al. Systematic profiling of poly (A) + transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 2015, 11, e1005166. [Google Scholar] [CrossRef]
- Powers, K.T.; Szeto, J.A.; Schaffitzel, C. New insights into no-go, non-stop and nonsense-mediated mRNA decay complexes. Curr. Opin. Struct. Biol. 2020, 65, 110–118. [Google Scholar] [CrossRef]
- Chiabudini, M.; Conz, C.; Reckmann, F.; Rospert, S. Ribosome-associated complex and Ssb are required for translational repression induced by polylysine segments within nascent chains. Mol. Cell Biol. 2012, 32, 4769–4779. [Google Scholar] [CrossRef] [Green Version]
- Taliaferro, J.M.; Vidaki, M.; Oliveira, R.; Olson, S.; Zhan, L.; Saxena, T.; Wang, E.T.; Graveley, B.R.; Gertler, F.B.; Swanson, M.S.; et al. Distal Alternative Last Exons Localize mRNAs to Neural Projections. Mol. Cell. 2016, 61, 821833. [Google Scholar] [CrossRef] [Green Version]
- Alt, F.W.; Bothwell, A.L.; Knapp, M.; Siden, E.; Mather, E.; Koshland, M.; Baltimore, D. Synthesis of secreted and membrane-bound immunoglobulin mu heavy chains is directed by mRNAs that differ at their 3′ ends. Cell 1980, 20, 293–301. [Google Scholar] [CrossRef]
- Rogers, J.; Early, P.; Carter, C.; Calame, K.; Bond, M.; Hood, L.; Wall, R. Two mRNAs with different 3′ ends encode membrane-bound and secreted forms of immunoglobulin mu chain. Cell 1980, 20, 303–312. [Google Scholar] [CrossRef]
- Early, P.; Rogers, J.; Davis, M.; Calame, K.; Bond, M.; Wall, R.; Hood, L. Two mRNAs can be produced from a single immunoglobulin mu gene by alternative RNA processing pathways. Cell 1980, 20, 313–319. [Google Scholar] [CrossRef]
- Davis, M.J.; Hanson, K.A.; Clark, F.; Fink, J.L.; Zhang, F.; Kasukawa, T.; Kai, C.; Kawai, J.; Carninci, P.; Hayashizaki, Y.; et al. Differential use of signal peptides and membrane domains is a common occurrence in the protein output of transcriptional units. PLoS Genet. 2006, 2, e46. [Google Scholar] [CrossRef] [Green Version]
- Singh, I.; Lee, S.H.; Sperling, A.S.; Samur, M.K.; Tai, Y.T.; Fulciniti, M.; Munshi, N.C.; Mayr, C.; Leslie, C.S. Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nat. Commun. 2018, 9, 1716. [Google Scholar] [CrossRef]
- Lee, S.H.; Singh, I.; Tisdale, S.; Abdel-Wahab, O.; Leslie, C.S.; Mayr, C. Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature 2018, 561, 127–131. [Google Scholar] [CrossRef]
- Dubbury, S.J.; Boutz, P.L.; Sharp, P.A. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 2018, 564, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Makarewich, C.A.; Olson, E.N. Mining for Micropeptides. Trends Cell Biol. 2017, 27, 685–696. [Google Scholar] [CrossRef]
- Thomas, C.P.; Andrews, J.I.; Liu, K.Z. Intronic polyadenylation signal sequences and alternate splicing generate human soluble Flt1 variants and regulate the abundance of soluble Flt1 in the placenta. FASEB J. 2007, 21, 3885–3895. [Google Scholar] [CrossRef]
- Vorlová, S.; Rocco, G.; Lefave, C.V.; Jodelka, F.M.; Hess, K.; Hastings, M.L.; Henke, E.; Cartegni, L. Induction of antagonistic soluble decoy receptor tyrosine kinases by intronic polyA activation. Mol. Cell 2011, 43, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, M.; Hewing, B.; Hui, J.; Zepp, A.; Baumann, G.; Bindereif, A.; Stangl, V.; Stangl, K. Alternative splicing in intron 13 of the human eNOS gene: A potential mechanism for regulating eNOS activity. FASEB J. 2007, 21, 1556–1564. [Google Scholar] [CrossRef]
- Mueller, A.A.; van Velthoven, C.T.; Fukumoto, K.D.; Cheung, T.H.; Rando, T.A. Intronic polyadenylation of PDGFRα in resident stem cells attenuates muscle fibrosis. Nature 2016, 540, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Stump, M.R.; Nguyen, R.T.; Drgastin, R.H.; Search, D.; Gong, Q.; Zhou, Z. Regulation of Kv11.1 Isoform Expression by Polyadenylate Binding Protein Nuclear 1. Int. J. Mol. Sci. 2021, 22, 863. [Google Scholar] [CrossRef]
- Xia, Z.; Donehower, L.A.; Cooper, T.A.; Neilson, J.R.; Wheeler, D.A.; Wagner, E.J.; Li, W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat. Commun. 2014, 5, 5274. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zheng, D.; Wei, L.; Ding, Q.; Tian, B. Regulation of Intronic Polyadenylation by PCF11 Impacts mRNA Expression of Long Genes. Cell Rep. 2019, 26, 2766–2778. [Google Scholar] [CrossRef] [Green Version]
- Di, C.; So, B.R.; Cai, Z.; Arai, C.; Duan, J.; Dreyfuss, G. U1 snRNP telescripting roles in transcription and its mechanism. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Woodbury, NY, USA, 2019; Volume 84, pp. 115–122. [Google Scholar] [CrossRef]
- Lang, A.J.; Mirski, S.E.; Cummings, H.J.; Yu, Q.; Gerlach, J.H.; Cole, S.P. Structural organization of the human TOP2A and TOP2B genes. Gene 1998, 221, 255–266. [Google Scholar] [CrossRef]
- Tsai-Pflugfelder, M.; Liu, L.F.; Liu, A.A.; Tewey, K.M.; Whang-Peng, J.; Knutsen, T.; Huebner, K.; Croce, C.M.; Wang, J.C. Cloning and sequencing of cDNA encoding human DNA topoisomerase II and localization of the gene to chromosome region 17q21-22. Proc. Natl. Acad. Sci. USA 1988, 85, 7177–7181. [Google Scholar] [CrossRef] [Green Version]
- Dong, K.C.; Berger, J.M. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature 2007, 450, 1201–1205. [Google Scholar] [CrossRef]
- Laponogov, I.; Pan, X.S.; Veselkov, D.A.; McAuley, K.E.; Fisher, L.M.; Sanderson, M.R. Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS ONE 2010, 5, e11338. [Google Scholar] [CrossRef]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The structure of DNA-bound human topoisomerase II a: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Harker, W.G.; Slade, D.L.; Dalton, W.S.; Meltzer, P.S.; Trent, J.M. Multidrug resistance in mitoxantrone-selected HL-60 leukemia cells in the absence of P-glycoprotein overexpression. Cancer Res. 1989, 49, 4542–4549. [Google Scholar] [PubMed]
- Harker, W.G.; Slade, D.L.; Parr, R.L.; Feldhoff, P.W.; Sullivan, D.M.; Holguin, M.H. Alterations in the topoisomerase II alpha gene, messenger RNA, and subcellular protein distribution as well as reduced expression of the DNA topoisomerase II beta enzyme in a mitoxantrone-resistant HL-60 human leukemia cell line. Cancer Res. 1995, 55, 1707–1716. [Google Scholar] [PubMed]
- Mirski, S.E.; Gerlach, J.H.; Cummings, H.J.; Zirngibl, R.; Greer, P.A.; Cole, S.P. Bipartite nuclear localization signals in the C terminus of human topoisomerase II alpha. Exp. Cell Res. 1997, 237, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Mirski, S.E.; Gerlach, J.H.; Cole, S.P. Sequence determinants of nuclear localization in the alpha and beta isoforms of human topoisomerase II. Exp. Cell Res. 1999, 251, 329–339. [Google Scholar] [CrossRef]
- Ritke, M.K.; Yalowich, J.C. Altered gene expression in human leukemia K562 cells selected for resistance to etoposide. Biochem. Pharmacol. 1993, 46, 2007–2020. [Google Scholar] [CrossRef]
- Ritke, M.K.; Roberts, D.; Allan, W.P.; Raymond, J.; Bergoltz, V.V.; Yalowich, J.C. Altered stability of etoposide-induced topoisomerase II-DNA complexes in resistant human leukaemia K562 cells. Br. J. Cancer 1994, 69, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Frère, V.; Sourgen, F.; Monnot, M.; Troalen, F.; Fermandjian, S. A peptide fragment of human DNA topoisomerase II alpha forms a stable coiled-coil structure in solution. J. Biol. Chem. 1995, 270, 17502–17507. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.M.; Gamblin, S.J.; Harrison, S.C.; Wang, J.C. Structure and mechanism of DNA topoisomerase II. Nature 1996, 379, 225–232. [Google Scholar] [CrossRef]
- Frère-Gallois, V.; Krebs, D.; Scala, D.; Troalen, F.; Fermandjian, S. Peptide fragments of DNA topoisomerase II with helix-forming and coiled-coil-forming properties act as inhibitors of the enzyme. Eur. J. Biochem. 1997, 249, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Kroll, D.J. Homologous and heterologous protein-protein interactions of human DNA topoisomerase IIalpha. Arch. Biochem. Biophys. 1997, 345, 175–184. [Google Scholar] [CrossRef]
- Bjergbaek, L.; Jensen, S.; Westergaard, O.; Andersen, A.H. Using a biochemical approach to identify the primary dimerization regions in human DNA topoisomerase IIalpha. J. Biol. Chem. 1999, 274, 26529–26536. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, L.P.; Roper, D.I.; Hammonds, T.R.; Maxwell, A. The N-terminal domain of human topoisomerase IIalpha is a DNA-dependent ATPase. Biochemistry 1998, 37, 16997–17004. [Google Scholar] [CrossRef]
- Campbell, S.; Maxwell, A. The ATP-operated clamp of human DNA topoisomerase IIalpha: Hyperstimulation of ATPase by “piggy-back” binding. J. Mol. Biol. 2002, 320, 171–188. [Google Scholar] [CrossRef]
- Hu, T.; Sage, H.; Hsieh, T.S. ATPase domain of eukaryotic DNA topoisomerase II. Inhibition of ATPase activity by the anti-cancer drug bisdioxopiperazine and ATP/ADP-induced dimerization. J. Biol. Chem. 2002, 277, 5944–5951. [Google Scholar] [CrossRef] [Green Version]
- Christie, M.; Chang, C.W.; Róna, G.; Smith, K.M.; Stewart, A.G.; Takeda, A.A.; Fontes, M.R.; Stewart, M.; Vértessy, B.G.; Forwood, J.K.; et al. Structural biology and regulation of protein import into the nucleus. J. Mol. Biol. 2016, 428, 2060–2090. [Google Scholar] [CrossRef]
- Hicks, M.J.; Mueller, W.F.; Shepard, P.J.; Hertel, K.J. Competing upstream 5′ splice sites enhance the rate of proximal splicing. Mol. Cell Biol. 2010, 30, 1878–1886. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.C.; Ou, A.C.; Park, J.; Yu, F.; Yu, B.; Lee, A.; Yang, G.; Zhou, A.; Benz, E.J., Jr. RBFOX2 promotes protein 4.1R exon 16 selection via U1 snRNP recruitment. Mol. Cell Biol. 2012, 32, 513–526. [Google Scholar] [CrossRef] [Green Version]
- Eckert, D.; Andrée, N.; Razanau, A.; Zock-Emmenthal, S.; Lützelberger, M.; Plath, S.; Schmidt, H.; Guerra-Moreno, A.; Cozzuto, L.; Ayté, J.; et al. Prp4 kinase grants the license to splice splice: Control of weak splice sites during spliceosome activation. PLoS Genet. 2016, 12, e1005768. [Google Scholar] [CrossRef]
- Wickramasinghe, V.O.; Gonzàlez-Porta, M.; Perera, D.; Bartolozzi, A.R.; Sibley, C.R.; Hallegger, M.; Ule, J.; Marioni, J.C.; Venkitaraman, A.R. Regulation of constitutive and alternative mRNA splicing across the human transcriptome by PRPF8 is determined by 5′ splice site strength. Genome Biol. 2015, 16, 201. [Google Scholar] [CrossRef] [Green Version]
- Gong, Q.; Stump, M.R.; Dunn, A.R.; Deng, V.; Zhou, Z. Alternative splicing and polyadenylation contribute to the generation of hERG1 C-terminal isoforms. J. Biol. Chem. 2010, 285, 32233–32241. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Ji, Z.; Pan, Z.; You, B.; Hoque, M.; Li, W.; Gunderson, S.I.; Tian, B. The Conserved Intronic Cleavage and Polyadenylation Site of CstF-77 Gene Imparts Control of 3′ End Processing Activity through Feedback Autoregulation and by U1 snRNP. PLoS Genet. 2013, 9, e1003613. [Google Scholar] [CrossRef] [Green Version]
- Yue, M.; Ogawa, Y. CRISPR/Cas9-mediated modulation of splicing efficiency reveals short splicing isoform of Xist RNA is sufficient to induce X-chromosome inactivation. Nucleic Acids Res. 2018, 46, e26. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPRCas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef]
- Cioe, L.; McNab, A.; Hubbell, H.R.; Meo, P.; Curtis, P.; Rovera, G. Differential expression of the globin genes in human leukemia K562(S) cells induced to differentiate by hemin or butyric acid. Cancer Res. 1981, 41, 237–243. [Google Scholar] [PubMed]
- Zhou, B.; Ho, S.S.; Greer, S.U.; Zhu, X.; Bell, J.M.; Arthur, J.G.; Spies, N.; Zhang, X.; Byeon, S.; Pattni, R.; et al. Comprehensive, integrated, and phased whole-genome analysis of the primary ENCODE cell line K562. Genome Res. 2019, 29, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Rhine, C.L.; Cygan, K.J.; Soemedi, R.; Maguire, S.; Murray, M.F.; Monaghan, S.F.; Fairbrother, W.G. Hereditary cancer genes are highly susceptible to splicing mutations. PLOS Genet. 2018, 14, e1007231. [Google Scholar] [CrossRef] [Green Version]
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-González, H.; Chai, S.; Wang, F.; et al. Systematic analysis of splice-site-creating mutations in cancer. Cell Rep. 2018, 23, 270–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, I.; Flotte, T.R.; Keeler, A.M. CRISPR/Cas-Dependent and Nuclease-Free In Vivo Therapeutic Gene Editing. Hum. Gene Ther. 2021, 32, 275–293. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elton, T.S.; Hernandez, V.A.; Carvajal-Moreno, J.; Wang, X.; Ipinmoroti, D.; Yalowich, J.C. Intronic Polyadenylation in Acquired Cancer Drug Resistance Circumvented by Utilizing CRISPR/Cas9 with Homology-Directed Repair: The Tale of Human DNA Topoisomerase IIα. Cancers 2022, 14, 3148. https://doi.org/10.3390/cancers14133148
Elton TS, Hernandez VA, Carvajal-Moreno J, Wang X, Ipinmoroti D, Yalowich JC. Intronic Polyadenylation in Acquired Cancer Drug Resistance Circumvented by Utilizing CRISPR/Cas9 with Homology-Directed Repair: The Tale of Human DNA Topoisomerase IIα. Cancers. 2022; 14(13):3148. https://doi.org/10.3390/cancers14133148
Chicago/Turabian StyleElton, Terry S., Victor A. Hernandez, Jessika Carvajal-Moreno, Xinyi Wang, Deborah Ipinmoroti, and Jack C. Yalowich. 2022. "Intronic Polyadenylation in Acquired Cancer Drug Resistance Circumvented by Utilizing CRISPR/Cas9 with Homology-Directed Repair: The Tale of Human DNA Topoisomerase IIα" Cancers 14, no. 13: 3148. https://doi.org/10.3390/cancers14133148