
The Novel Immune Checkpoint GPR56 Is Expressed on Tumor-Infiltrating Lymphocytes and Selectively Upregulated upon TCR Signaling

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Analysis of Single Cell mRNA Sequencing Data
2.2. Isolation of Peripheral Blood Mononuclear Cells (PBMCs)
2.3. Isolation of Tumor Infiltrated Lymphocytes (TILs) from Fresh Tumor Tissue
2.4. Multiparameter Flow Cytometry
2.5. Cell Lines and Transfectants
2.6. T Cell and TIL Activation Assay
2.7. In Vitro Migration Assay
2.8. Statistical Analysis
3. Results
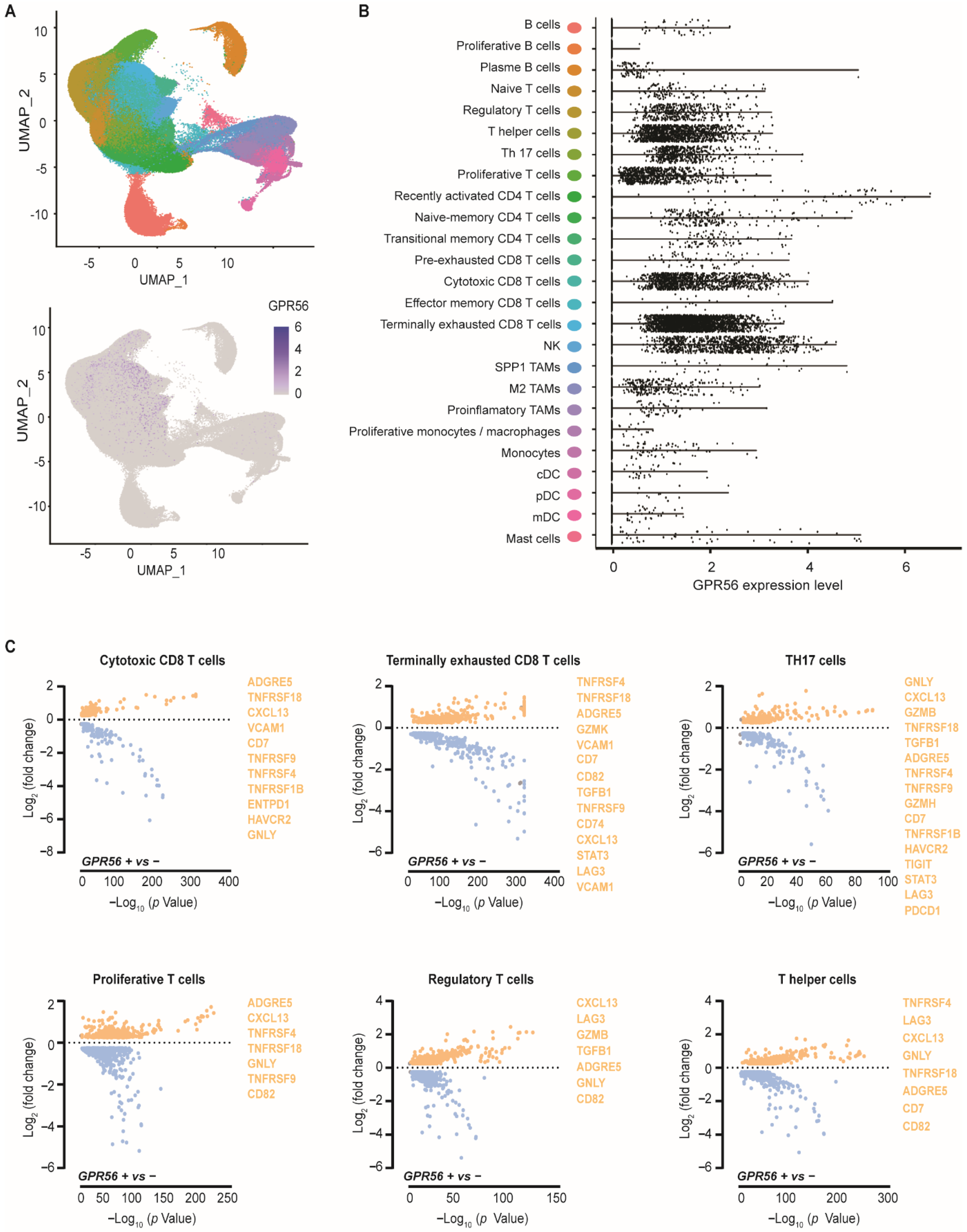
3.1. GPR56 Is Expressed in Cytotoxic and Terminally Exhausted CD8 TILs That Display a Tumor-Reactive Phenotype across Multiple Tumor Types
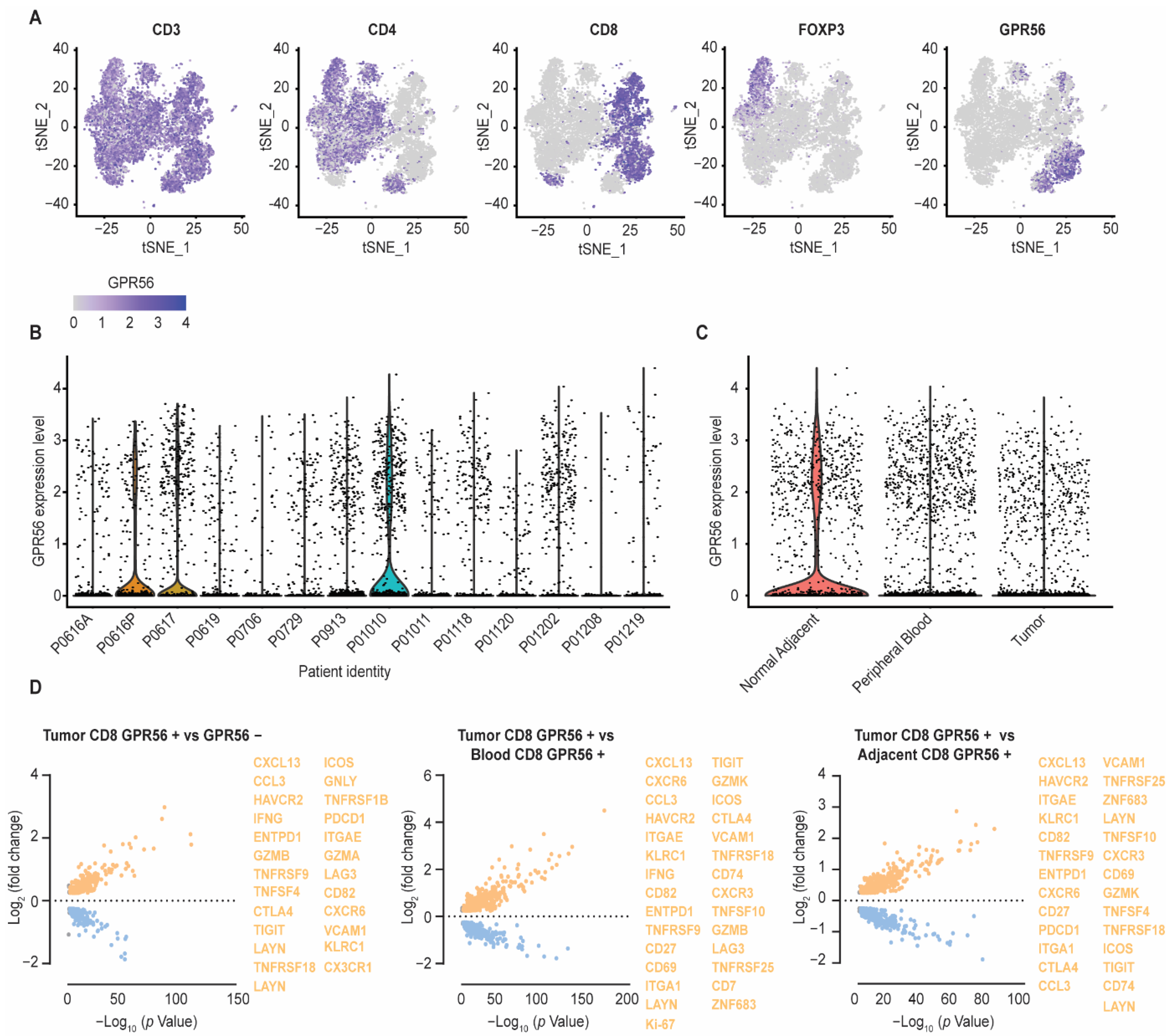
3.2. Compared to Blood and Adjacent Tissue, Tumor Localized GPR56-Positive CD8 T Cells Display a Distinct Gene Expression Profile with Upregulated Tumor-Reactive Markers
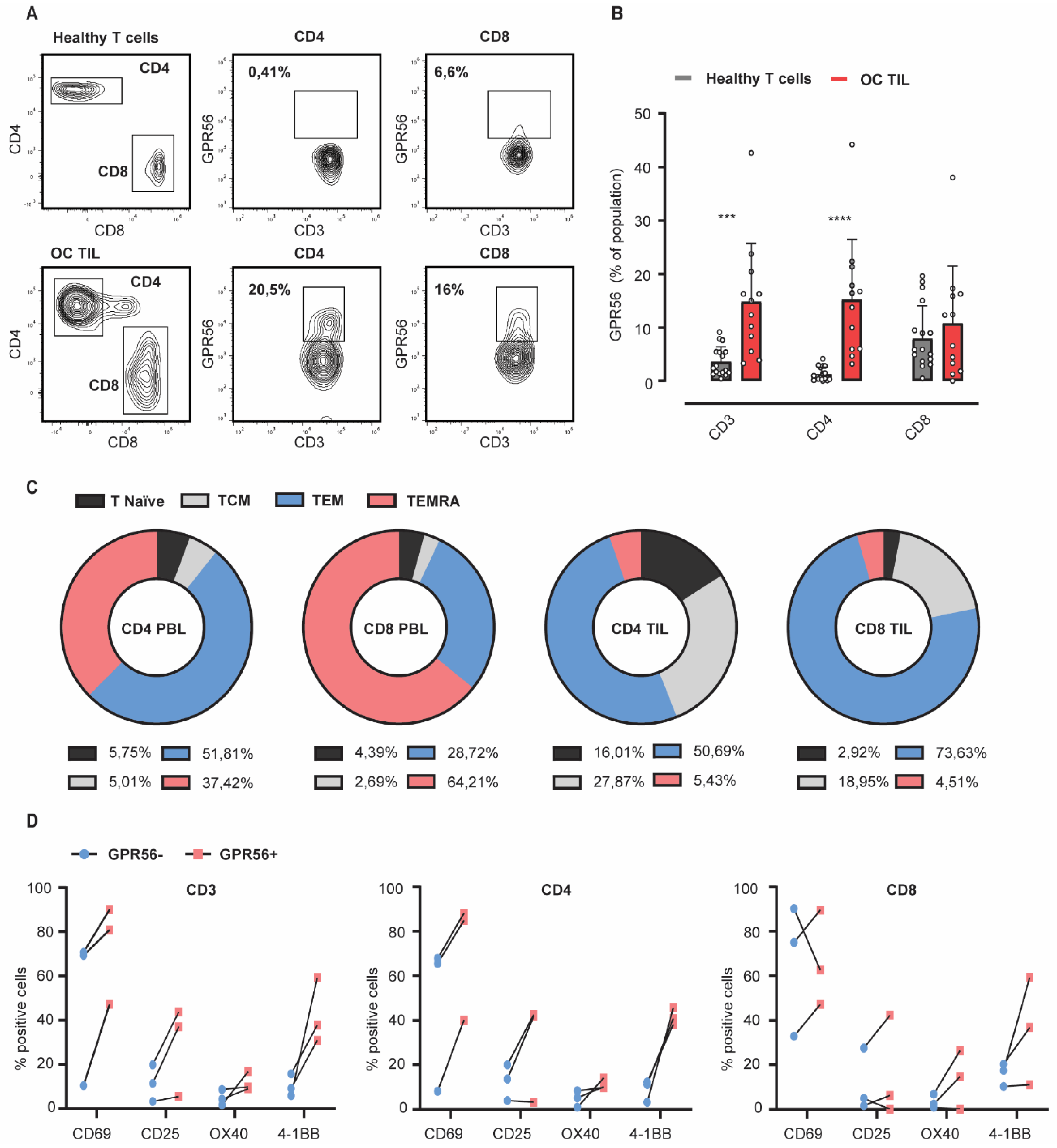
3.3. GPR56 Expression Is Elevated on Ovarian Cancer TILs Compared to Healthy Peripheral Blood Lymphocytes and Associates with an Effector Memory Phenotype
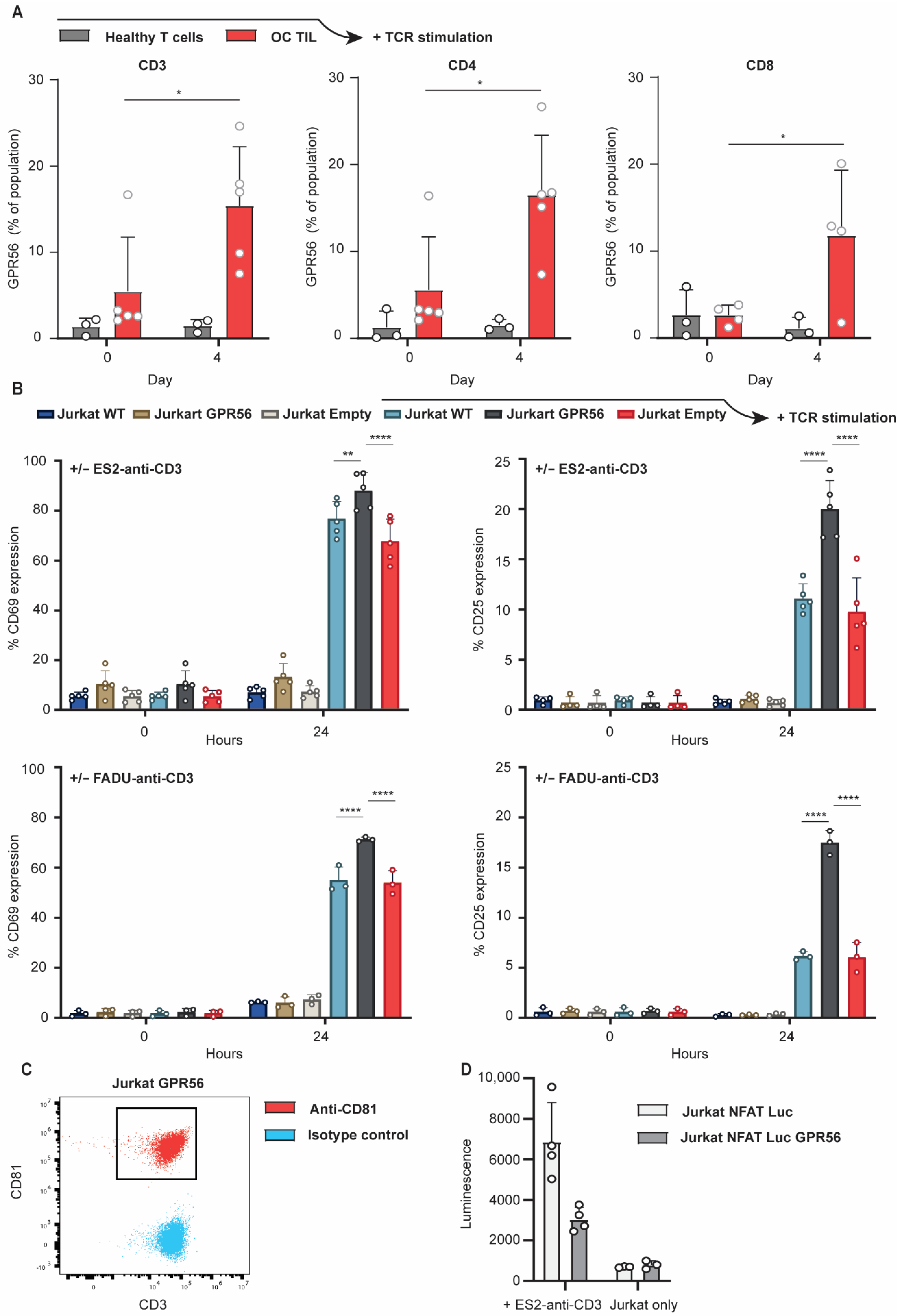
3.4. TCR Stimulation Upregulates GPR56 Expression on TILs and Enhances the Activation Status of T Cells
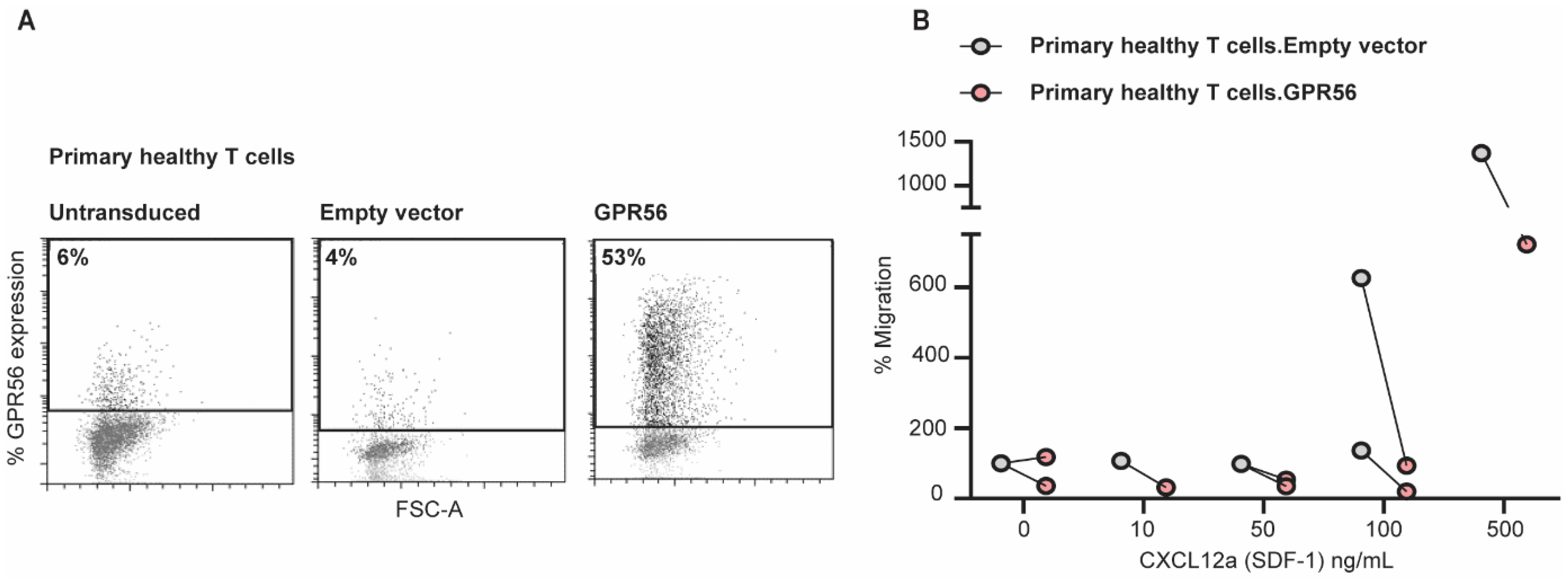
3.5. Ectopic GPR56 Expression Inhibits Migration of Primary Healthy T Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Jenkins, R.W.; Sullivan, R.J. Mechanisms of Resistance to Immune Checkpoint Blockade. Am. J. Clin. Dermatol. 2019, 20, 41–54. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Shmulevich, I. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Patel, M.R.; Pant, S.; Hamilton, E.P.; Lockhart, A.C.; Kelly, K.; Taylor, M.H. Avelumab (MSB0010718C; anti-PD-L1) in patients with recurrent/refractory ovarian cancer from the JAVELIN Solid Tumor phase Ib trial: Safety and clinical activity. J. Clin. Oncol. 2016, 34, 15. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Konishi, I. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef]
- Hodi, F.S.; Mihm, M.C.; Soiffer, R.J.; Haluska, F.G.; Butler, M.; Seiden, M.V.; Dranoff, G. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. USA 2003, 100, 4712–4717. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Ledermann, J.A. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Results from the phase II KEYNOTE-100 study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef]
- Polk, A.; Svane, I.M.; Andersson, M.; Nielsen, D. Checkpoint inhibitors in breast cancer—Current status. Cancer Treat. Rev. 2018, 63, 122–134. [Google Scholar] [CrossRef]
- Young, K.; Hughes, D.J.; Cunningham, D.; Starling, N. Immunotherapy and pancreatic cancer: Unique challenges and potential opportunities. Ther. Adv. Med. Oncol. 2018, 10, 1758835918816281. [Google Scholar] [CrossRef] [Green Version]
- Keane, C.; Law, S.C.; Gould, C.; Birch, S.; Sabdia, M.B.; Merida de Long, L.; Gandhi, M.K. LAG3: A novel immune checkpoint expressed by multiple lymphocyte subsets in diffuse large B-cell lymphoma. Blood Adv. 2020, 4, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Onco. Targets Ther. 2020, 11, 7005–7009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamm, H.; Oliveira-Ferrer, L.; Grossjohann, E.M.; Muschhammer, J.; Thaden, V.; Brauneck, F.; Wellbrock, J. Targeting the TIGIT-PVR immune checkpoint axis as novel therapeutic option in breast cancer. Oncoimmunology 2019, 8, e1674605. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Mo, S.; Yu, S.; Zhou, Y.; Zhang, M.; Chen, J.; Xiang, Y. Expression of the immune checkpoint VISTA in breast cancer. Cancer Immunol. Immunother. 2020, 69, 1437–1446. [Google Scholar] [CrossRef]
- Krijgsman, D.; Roelands, J.; Hendrickx, W.; Bedognetti, D.; Kuppen, P.J.K. HLA-G: A New Immune Checkpoint in Cancer? Int. J. Mol. Sci. 2020, 21, 4528. [Google Scholar] [CrossRef]
- Van Hall, T.; Andre, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Yan, H.; Qiu, W.; Koehne de Gonzalez, A.K.; Wei, J.S.; Tu, M.; Xi, C.H.; Su, G.H. HHLA2 is a novel immune checkpoint protein in pancreatic ductal adenocarcinoma and predicts post-surgical survival. Cancer Lett. 2019, 442, 333–340. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Tan, L.J.C.D. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [Green Version]
- Aubert, N.; Brunel, S.; Olive, D.; Marodon, G.J.B. TNFRSF14 (HVEM) is a novel immune checkpoint for cancer immunotherapy in humanized mice. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Bae, B.I.; Tietjen, I.; Atabay, K.D.; Evrony, G.D.; Johnson, M.B.; Asare, E.; Walsh, C.A. Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science 2014, 343, 764–768. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Jin, Z.; Koirala, S.; Bu, L.; Xu, L.; Hynes, R.O.; Piao, X. GPR56 regulates pial basement membrane integrity and cortical lamination. J. Neurosci. 2008, 28, 5817–5826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belzeaux, R.; Gorgievski, V.; Fiori, L.M.; Lopez, J.P.; Grenier, J.; Lin, R.; Richard-Devantoy, S.J.N.C. GPR56/ADGRG1 is associated with response to antidepressant treatment. Nat. Commun. 2020, 11, 1635. [Google Scholar] [CrossRef] [PubMed]
- Maglitto, A.; Mariani, S.A.; de Pater, E.; Rodriguez-Seoane, C.; Vink, C.S.; Piao, X.; Dzierzak, E. Unexpected redundancy of Gpr56 and Gpr97 during hematopoietic cell development and differentiation. Blood Adv. 2021, 5, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Fan, J.; Yang, J.; Zhu, G.-Z.J.M. Characterization of GPR56 protein and its suppressed expression in human pancreatic cancer cells. Mol. Cell. Biochem. 2008, 308, 133–139. [Google Scholar] [CrossRef]
- Shashidhar, S.; Lorente, G.; Nagavarapu, U.; Nelson, A.; Kuo, J.; Cummins, J.; Foehr, E.D. GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene 2005, 24, 1673–1682. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Begum, S.; Hearn, J.D.; Hynes, R.O. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 9023–9028. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.R.; Kang, D.H.; Kuk, J.C.; Kim, T.H.; Shin, E.J.; Ahn, T.S.; Kim, N.K. Prognostic impact of GPR56 in patients with colorectal cancer. Neoplasma 2021, 68, 580–589. [Google Scholar] [CrossRef]
- Pabst, C.; Bergeron, A.; Lavallee, V.P.; Yeh, J.; Gendron, P.; Norddahl, G.L.; Barabe, F. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood 2016, 127, 2018–2027. [Google Scholar] [CrossRef] [Green Version]
- Chang, G.W.; Hsiao, C.C.; Peng, Y.M.; Vieira Braga, F.A.; Kragten, N.A.; Remmerswaal, E.B.; Hamann, J. The Adhesion G Protein-Coupled Receptor GPR56/ADGRG1 Is an Inhibitory Receptor on Human NK Cells. Cell Rep. 2016, 15, 1757–1770. [Google Scholar] [CrossRef] [Green Version]
- Della Chiesa, M.; Falco, M.; Parolini, S.; Bellora, F.; Petretto, A.; Romeo, E.; Vitale, M. GPR56 as a novel marker identifying the CD56dull CD16+ NK cell subset both in blood stream and in inflamed peripheral tissues. Int. Immunol. 2010, 22, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Janks, L.; Habiel, D.; Litvak, V.; Lin, L.-L.; Kwon, H. GPR56 inhibits chemotaxis of human Natural Killer and TEMRA lymphocytes. J. Immunol. 2020, 204, 220–222. [Google Scholar]
- Peng, Y.M.; van de Garde, M.D.; Cheng, K.F.; Baars, P.A.; Remmerswaal, E.B.; van Lier, R.A.; Hamann, J. Specific expression of GPR56 by human cytotoxic lymphocytes. J. Leukoc. Biol. 2011, 90, 735–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, K.L.; Schlickeiser, S.; Vogt, K.; Boes, D.; Stanko, K.; Appelt, C.; Sawitzki, B. Killer-like receptors and GPR56 progressive expression defines cytokine production of human CD4(+) memory T cells. Nat. Commun. 2019, 10, 2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Zhang, Z. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef]
- Nieto, P.; Elosua-Bayes, M.; Trincado, J.L.; Marchese, D.; Massoni-Badosa, R.; Salvany, M.; Heyn, H. A Single-Cell Tumor Immune Atlas for Precision Oncology. Genome Res. 2021, 31, 1913–1926. [Google Scholar] [CrossRef]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Gottardo, R. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 278. [Google Scholar] [CrossRef] [Green Version]
- Human Tissue and Medical Research: Code of Conduct for Responsible Use. 2011. Available online: https://www.federa.org/sites/default/files/images/print_version_code_of_conduct_english.pdf (accessed on 17 June 2022).
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; de Miranda, N.F.; Weinberg, A.D. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 2724. [Google Scholar] [CrossRef] [Green Version]
- Eiva, M.A.; Omran, D.K.; Chacon, J.; Powell, D.J. Systematic analysis of CD39, CD103, CD137 and PD-1 as biomarkers for naturally occurring tumor antigen-specific TILs. Eur. J. Immunol. 2022, 52, 96–108. [Google Scholar] [CrossRef]
- Workel, H.H.; van Rooij, N.; Plat, A.; Spierings, D.C.J.; Fehrmann, R.S.N.; Nijman, H.W.; de Bruyn, M. Transcriptional Activity and Stability of CD39+CD103+CD8+ T Cells in Human High-Grade Endometrial Cancer. Int. J. Mol. Sci. 2020, 21, 3770. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, Z.J.A. Understanding tumor-infiltrating lymphocytes by single cell RNA sequencing. Adv. Immunol. 2019, 144, 217–245. [Google Scholar] [PubMed]
- Tian, Y.; Babor, M.; Lane, J.; Schulten, V.; Patil, V.S.; Seumois, G.; Peters, B. Unique phenotypes and clonal expansions of human CD4 effector memory T cells re-expressing CD45RA. Nat. Commun. 2017, 8, 1473. [Google Scholar] [CrossRef]
- Truong, K.-L.; Schlickeiser, S.; Vogt, K.; Boës, D.; Schumann, J.; Appelt, C.; Sawitzki, B. Progressive change in expression of killer-like receptorsr and GPR56 expression defines the cytokine production potential of human CD4+ T memory cells. bioRxiv 2017, 10, 2263. [Google Scholar] [CrossRef]
- Wanjalla, C.N.; McDonnell, W.J.; Barnett, L.; Simmons, J.D.; Furch, B.D.; Lima, M.C.; Koethe, J.R. Adipose Tissue in Persons With HIV Is Enriched for CD4(+) T Effector Memory and T Effector Memory RA(+) Cells, Which Show Higher CD69 Expression and CD57, CX3CR1, GPR56 Co-expression With Increasing Glucose Intolerance. Front. Immunol. 2019, 10, 408. [Google Scholar] [CrossRef] [Green Version]
- Bottcher, J.P.; Beyer, M.; Meissner, F.; Abdullah, Z.; Sander, J.; Hochst, B.; Knolle, P.A. Functional classification of memory CD8(+) T cells by CX3CR1 expression. Nat. Commun. 2015, 6, 8306. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, T.; Sakata, K.; Yoshizaki, K.; Tago, K.; Mizuno, N.; Itoh, H. Orphan G protein-coupled receptor GPR56 regulates neural progenitor cell migration via a G alpha 12/13 and Rho pathway. J. Biol. Chem. 2008, 283, 14469–14478. [Google Scholar] [CrossRef] [Green Version]
- Singer, K.; Luo, R.; Jeong, S.J.; Piao, X. GPR56 and the developing cerebral cortex: Cells, matrix, and neuronal migration. Mol. Neurobiol. 2013, 47, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.Y.; Peng, Y.M.; Juang, H.H.; Chen, T.C.; Pan, H.L.; Chang, G.W.; Lin, H.H. GPR56/ADGRG1 Activation Promotes Melanoma Cell Migration via NTF Dissociation and CTF-Mediated Galpha12/13/RhoA Signaling. J. Invest. Dermatol. 2017, 137, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, T.; Zhang, S.; Posey, T.A.; Jacob, J.; Wu, L.; Yu, W.; Carmon, K.S. Anti-GPR56 monoclonal antibody potentiates GPR56-mediated Src-Fak signaling to modulate cell adhesion. J. Biol. Chem. 2021, 296, 100261. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilemjian, V.; Vlaming, M.R.; Álvarez Freile, J.; Huls, G.; De Bruyn, M.; Bremer, E. The Novel Immune Checkpoint GPR56 Is Expressed on Tumor-Infiltrating Lymphocytes and Selectively Upregulated upon TCR Signaling. Cancers 2022, 14, 3164. https://doi.org/10.3390/cancers14133164
Bilemjian V, Vlaming MR, Álvarez Freile J, Huls G, De Bruyn M, Bremer E. The Novel Immune Checkpoint GPR56 Is Expressed on Tumor-Infiltrating Lymphocytes and Selectively Upregulated upon TCR Signaling. Cancers. 2022; 14(13):3164. https://doi.org/10.3390/cancers14133164
Chicago/Turabian StyleBilemjian, Vrouyr, Martijn R. Vlaming, Jimena Álvarez Freile, Gerwin Huls, Marco De Bruyn, and Edwin Bremer. 2022. "The Novel Immune Checkpoint GPR56 Is Expressed on Tumor-Infiltrating Lymphocytes and Selectively Upregulated upon TCR Signaling" Cancers 14, no. 13: 3164. https://doi.org/10.3390/cancers14133164