
A Novel Role of SMG1 in Cholesterol Homeostasis That Depends Partially on p53 Alternative Splicing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Cell Viability Analysis
2.3. Western Blots
2.4. Real-Time PCR and miRNA Assay
2.5. Transcriptome and Analysis
2.6. Cholesterol Staining and Analysis
3. Results
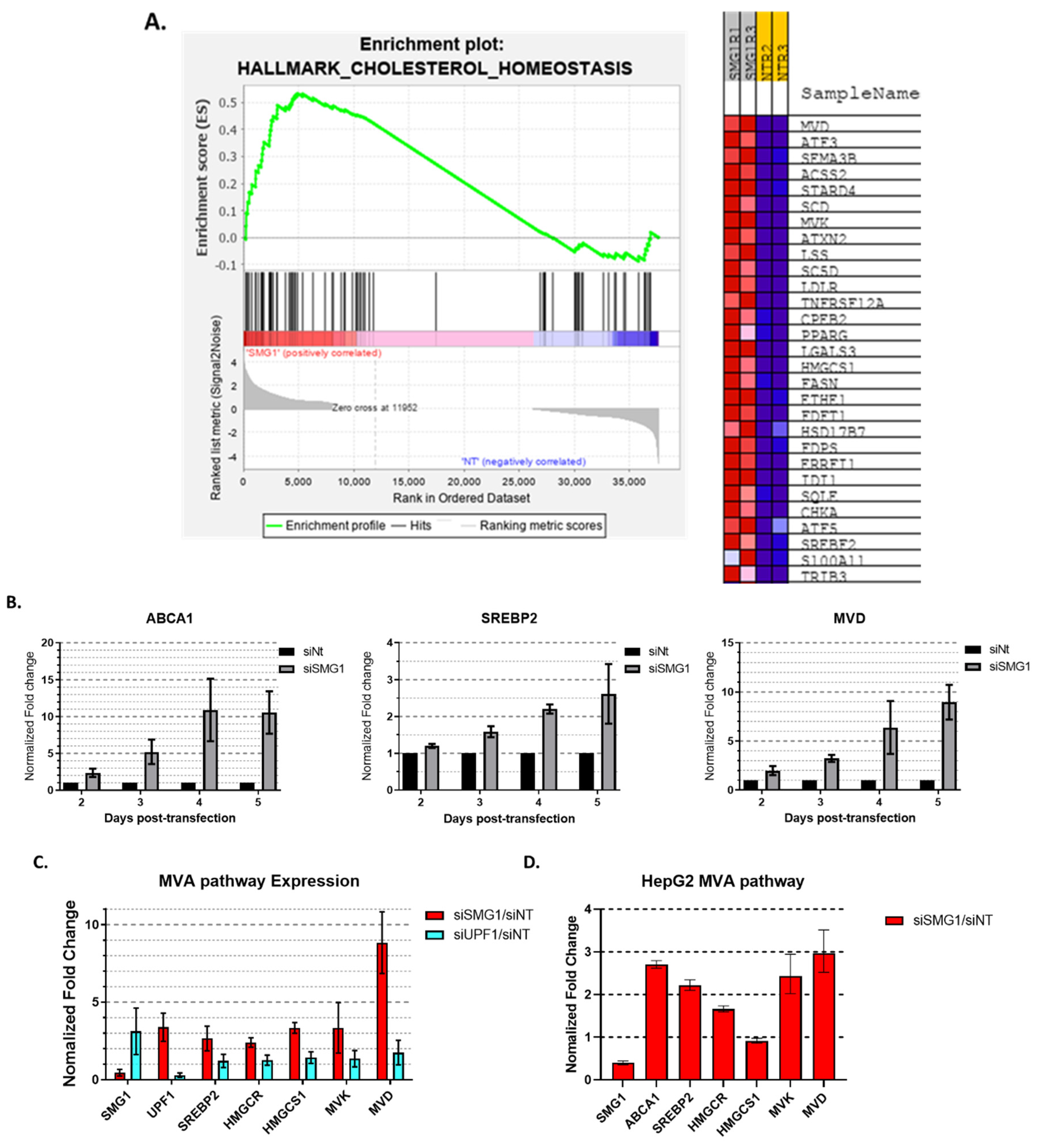
3.1. SMG1 Is an Unrecognized Cholesterol Metabolism Regulator
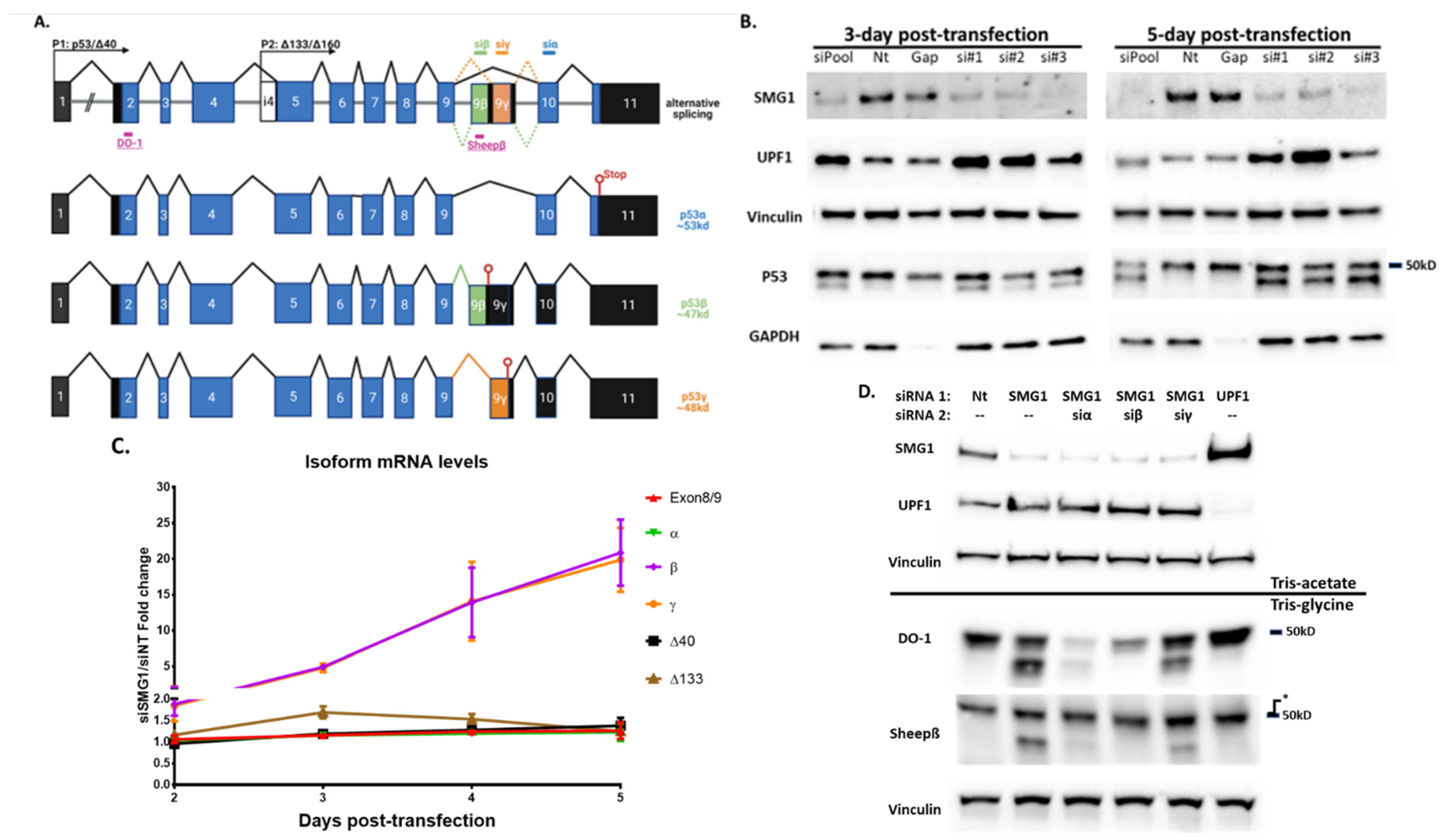
3.2. Loss of SMG1 Alternates p53 Isoform Splicing
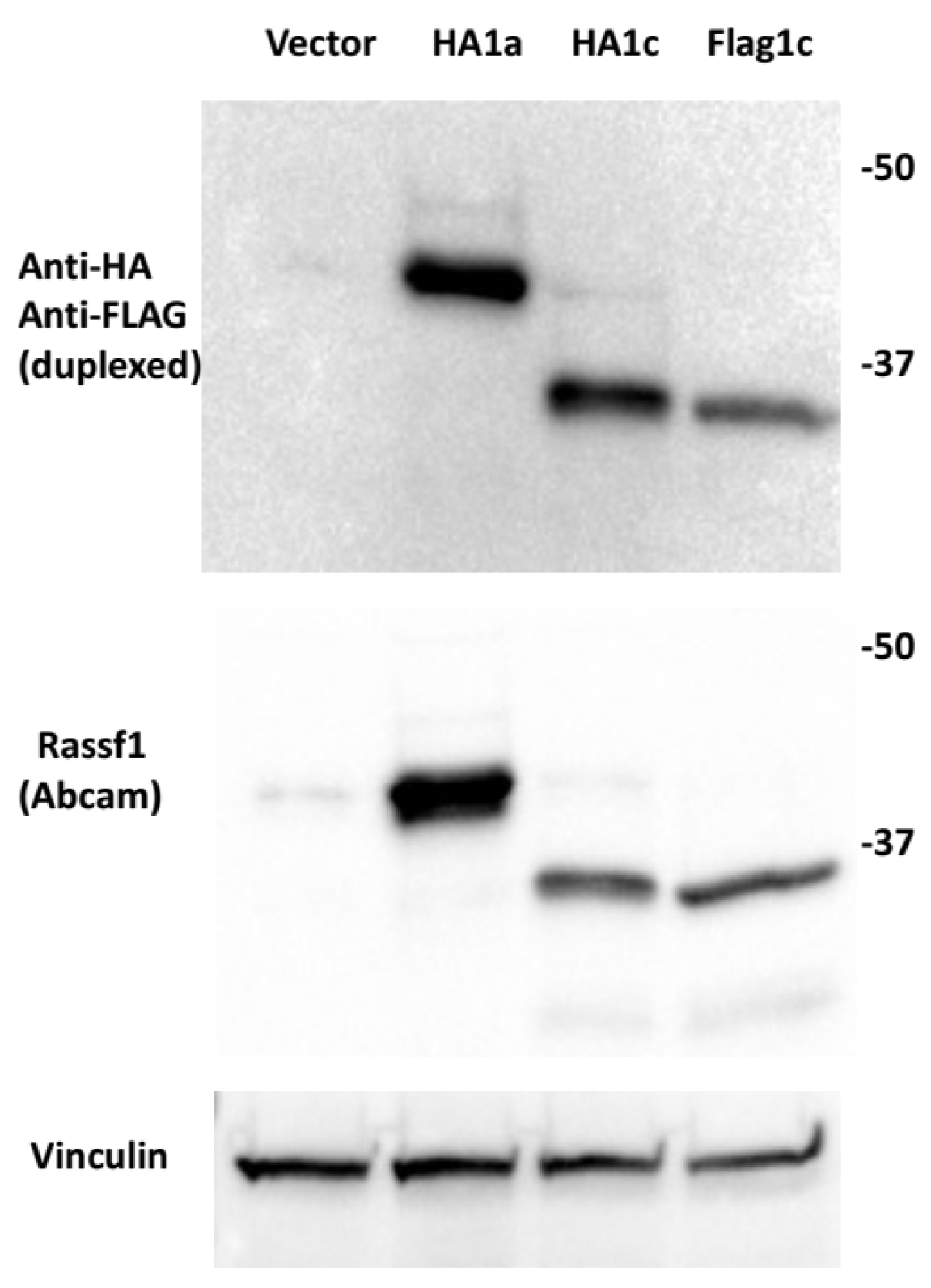
3.3. P53 Isoforms Modulate the Expression of ABCA1 Differently with No Effects on SREBP2 or MVD
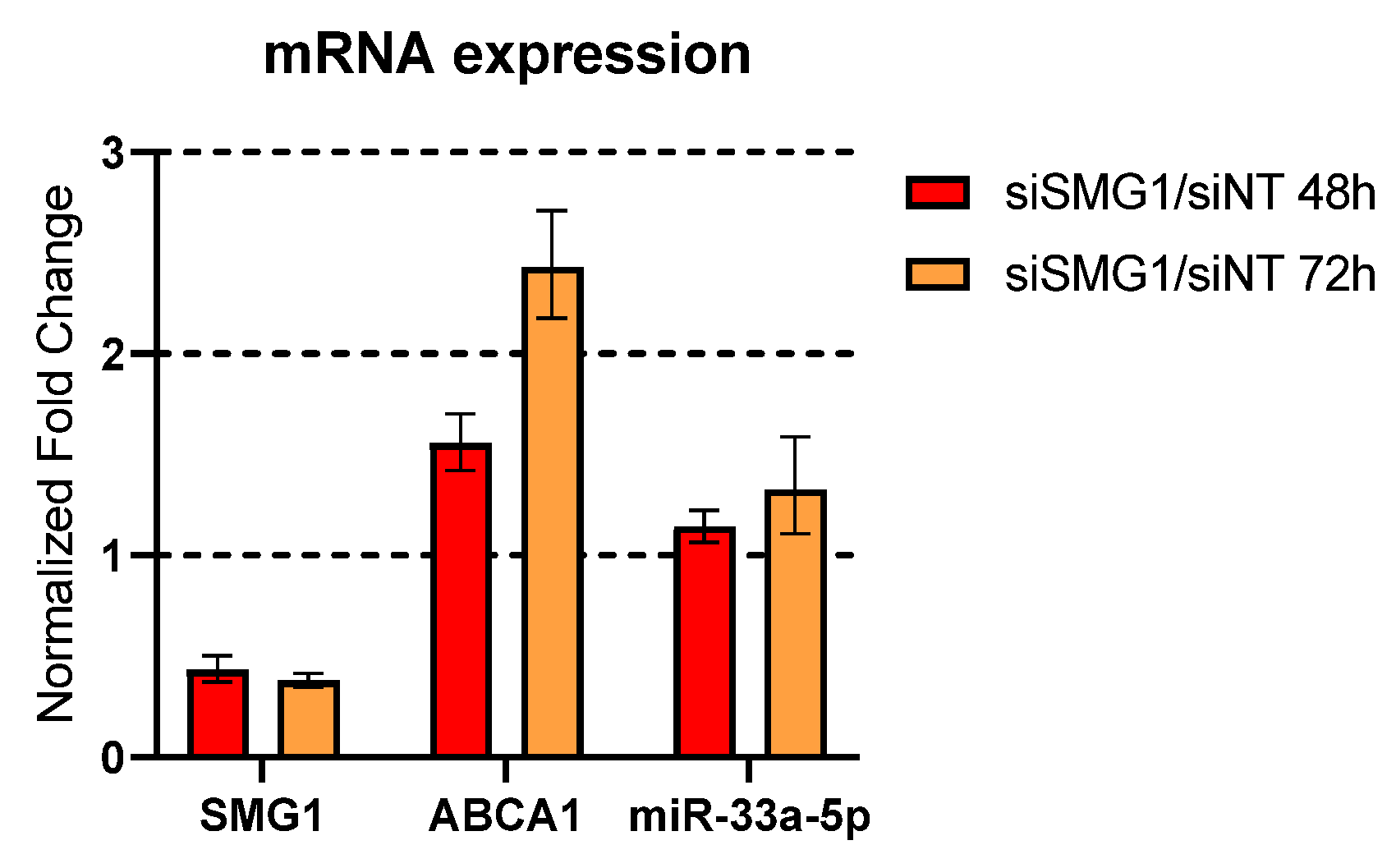
3.4. miR-33a, the Canonical ABCA1 Inhibitor Is Not Perturbated by Loss of SMG1
3.5. Loss of SMG1 Increased Intracellular Cholesterol Level
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



References
- Lloyd, J.P.; Davies, B. SMG1 is an ancient nonsense-mediated mRNA decay effector. Plant J. 2013, 76, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Brumbaugh, K.M.; Otterness, D.M.; Geisen, C.; Oliveira, V.; Brognard, J.; Li, X.; Lejeune, F.; Tibbetts, R.S.; Maquat, L.E.; Abraham, R.T. The mRNA surveillance protein hSMG-1 functions in genotoxic stress response pathways in mammalian cells. Mol. Cell 2004, 14, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Gewandter, J.S.; Bambara, R.A.; O’Reilly, M.A. The RNA surveillance protein SMG1 activates p53 in response to DNA double-strand breaks but not exogenously oxidized mRNA. Cell Cycle 2011, 10, 2561–2567. [Google Scholar] [CrossRef] [Green Version]
- Gubanova, E.; Issaeva, N.; Gokturk, C.; Djureinovic, T.; Helleday, T. SMG-1 suppresses CDK2 and tumor growth by regulating both the p53 and Cdc25A signaling pathways. Cell Cycle 2013, 12, 3770–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehen, S.C.; Staversky, R.J.; Bambara, R.A.; Keng, P.C.; O’Reilly, M.A. hSMG-1 and ATM sequentially and independently regulate the G1 checkpoint during oxidative stress. Oncogene 2008, 27, 4065–4074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, S.-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A. Role of SMG-1-mediated Upf1 phosphorylation in mammalian nonsense-mediated mRNA decay. Genes Cells 2013, 18, 161–175. [Google Scholar] [CrossRef]
- Yamashita, A.; Izumi, N.; Kashima, I.; Ohnishi, T.; Saari, B.; Katsuhata, Y.; Muramatsu, R.; Morita, T.; Iwamatsu, A.; Hachiya, T.; et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 2009, 23, 1091–1105. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Jacobson, A. Nonsense-Mediated mRNA Decay: Degradation of Defective Transcripts Is Only Part of the Story. Annu Rev. Genet. 2015, 49, 339–366. [Google Scholar] [CrossRef] [Green Version]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Gubanova, E.; Brown, B.; Ivanov, S.V.; Helleday, T.; Mills, G.B.; Yarbrough, W.G.; Issaeva, N. Downregulation of SMG-1 in HPV-positive head and neck squamous cell carcinoma due to promoter hypermethylation correlates with improved survival. Clin. Cancer Res. 2012, 18, 1257–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, T.L.; Ho, U.; Luff, J.; Lee, C.S.; Apte, S.H.; MacDonald, K.P.; Raggat, L.J.; Pettit, A.R.; Morrow, C.A.; Waters, M.J.; et al. Smg1 haploinsufficiency predisposes to tumor formation and inflammation. Proc. Natl. Acad. Sci. USA 2013, 110, E285–E294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalsamy, A.; Bennett, E.M.; Shi, M.; Zhang, W.G.; Bard, J.; Yu, K. Identification of pyrimidine derivatives as hSMG-1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 6636–6641. [Google Scholar] [CrossRef] [PubMed]
- Matlashewski, G.; Lamb, P.; Pim, D.; Peacock, J.; Crawford, L.; Benchimol, S. Isolation and characterization of a human p53 cDNA clone: Expression of the human p53 gene. EMBO J. 1984, 3, 3257–3262. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Rotter, V. Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc. Natl. Acad. Sci. USA 1985, 82, 790–794. [Google Scholar] [CrossRef] [Green Version]
- Bourdon, J.C. p53 and its isoforms in cancer. Br. J. Cancer 2007, 97, 277–282. [Google Scholar] [CrossRef]
- Ghosh, A.; Stewart, D.; Matlashewski, G. Regulation of human p53 activity and cell localization by alternative splicing. Mol. Cell. Biol. 2004, 24, 7987–7997. [Google Scholar] [CrossRef] [Green Version]
- Courtois, S.; Caron de Fromentel, C.; Hainaut, P. p53 protein variants: Structural and functional similarities with p63 and p73 isoforms. Oncogene 2004, 23, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef]
- Joruiz, S.M.; Bourdon, J.C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowen, L.E.; Tang, Y. Identification of nonsense-mediated mRNA decay pathway as a critical regulator of p53 isoform beta. Sci. Rep. 2017, 7, 17535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudikote, J.P.; Cascone, T.; Poteete, A.; Sitthideatphaiboon, P.; Wu, Q.; Morikawa, N.; Zhang, F.; Peng, S.; Tong, P.; Li, L.; et al. Inhibition of nonsense-mediated decay rescues p53beta/gamma isoform expression and activates the p53 pathway in MDM2-overexpressing and select p53-mutant cancers. J. Biol. Chem. 2021, 297, 101163. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Crutchley, J.; Zhang, D.D.; Owzar, K.; Kastan, M.B. Identification of a DNA Damage-Induced Alternative Splicing Pathway That Regulates p53 and Cellular Senescence Markers. Cancer Discov. 2017, 7, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53beta, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.T.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e519. [Google Scholar] [CrossRef] [Green Version]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Vedhachalam, C.; Duong, P.T.; Nickel, M.; Nguyen, D.; Dhanasekaran, P.; Saito, H.; Rothblat, G.H.; Lund-Katz, S.; Phillips, M.C. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J. Biol. Chem. 2007, 282, 25123–25130. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Li, L.; Li, W.; Yang, F.; Zhang, Z.; Liu, Z.; Du, W. p53 transcriptionally regulates SQLE to repress cholesterol synthesis and tumor growth. EMBO Rep. 2021, 22, e52537. [Google Scholar] [CrossRef]
- Bongiorno, R.; Colombo, M.P.; Lecis, D. Deciphering the nonsense-mediated mRNA decay pathway to identify cancer cell vulnerabilities for effective cancer therapy. J. Exp. Clin. Cancer Res. 2021, 40, 376. [Google Scholar] [CrossRef]
- Nogueira, G.; Fernandes, R.; Garcia-Moreno, J.F.; Romao, L. Nonsense-mediated RNA decay and its bipolar function in cancer. Mol. Cancer 2021, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, D.; Qin, X.; Owzar, K.; McCann, J.J.; Kastan, M.B. DNA-Damage-Induced Alternative Splicing of p53. Cancers 2021, 13, 251. [Google Scholar] [CrossRef] [PubMed]
- Sumitra Deb, S.P.D. p53 Protocols: Methods in Molecular Biology; Humana: Louisville, KY, USA, 2013; Volume 962. [Google Scholar]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Newton, J.; Palladino, E.N.D.; Weigel, C.; Maceyka, M.; Graler, M.H.; Senkal, C.E.; Enriz, R.D.; Marvanova, P.; Jampilek, J.; Lima, S.; et al. Targeting defective sphingosine kinase 1 in Niemann-Pick type C disease with an activator mitigates cholesterol accumulation. J. Biol. Chem. 2020, 295, 9121–9133. [Google Scholar] [CrossRef]
- Camus, S.; Menendez, S.; Fernandes, K.; Kua, N.; Liu, G.; Xirodimas, D.P.; Lane, D.P.; Bourdon, J.C. The p53 isoforms are differentially modified by Mdm2. Cell Cycle 2012, 11, 1646–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laubach, K.; Zhang, J.; Chen, X. The p53 Family: A Role in Lipid and Iron Metabolism. Front. Cell Dev. Biol. 2021, 9, 715974. [Google Scholar] [CrossRef]
- Parrales, A.; Iwakuma, T. p53 as a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci. 2016, 17, 2074. [Google Scholar] [CrossRef]
- Marquart, T.J.; Allen, R.M.; Ory, D.S.; Baldan, A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc. Natl. Acad. Sci. USA 2010, 107, 12228–12232. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Merchan, A.; Cerrato, C.; Luengo, G.; Dominguez, O.; Piris, M.A.; Serrano, M.; Gonzalez, S. miR-33-mediated downregulation of p53 controls hematopoietic stem cell self-renewal. Cell Cycle 2010, 9, 3277–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi-Shoushtari, S.H.; Kristo, F.; Li, Y.; Shioda, T.; Cohen, D.E.; Gerszten, R.E.; Naar, A.M. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 2010, 328, 1566–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayner, K.J.; Suarez, Y.; Davalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernandez-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaar, Y.G.; Reeves, M.E. RASSF1C regulates miR-33a and EMT marker gene expression in lung cancer cells. Oncotarget 2019, 10, 123–132. [Google Scholar] [CrossRef]
- Burbee, D.G.; Forgacs, E.; Zochbauer-Muller, S.; Shivakumar, L.; Fong, K.; Gao, B.; Randle, D.; Kondo, M.; Virmani, A.; Bader, S.; et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J. Natl. Cancer Inst. 2001, 93, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Dammann, R.; Schagdarsurengin, U.; Strunnikova, M.; Rastetter, M.; Seidel, C.; Liu, L.; Tommasi, S.; Pfeifer, G.P. Epigenetic inactivation of the Ras-association domain family 1 (RASSF1A) gene and its function in human carcinogenesis. Histol. Histopathol. 2003, 18, 665–677. [Google Scholar] [CrossRef]
- Gholkar, A.A.; Cheung, K.; Williams, K.J.; Lo, Y.C.; Hamideh, S.A.; Nnebe, C.; Khuu, C.; Bensinger, S.J.; Torres, J.Z. Fatostatin Inhibits Cancer Cell Proliferation by Affecting Mitotic Microtubule Spindle Assembly and Cell Division. J. Biol. Chem. 2016, 291, 17001–17008. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Machamer, C.E.; Espenshade, P.J. Fatostatin blocks ER exit of SCAP but inhibits cell growth in a SCAP-independent manner. J. Lipid Res. 2016, 57, 1564–1573. [Google Scholar] [CrossRef] [Green Version]
- Karam, R.; Lou, C.H.; Kroeger, H.; Huang, L.; Lin, J.H.; Wilkinson, M.F. The unfolded protein response is shaped by the NMD pathway. EMBO Rep. 2015, 16, 599–609. [Google Scholar] [CrossRef]
- Marcel, V.; Fernandes, K.; Terrier, O.; Lane, D.P.; Bourdon, J.C. Modulation of p53beta and p53gamma expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 2014, 21, 1377–1387. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Philantrope, F.; Diot, A.; Bourdon, J.-C.; Thompson, P. A Novel Role of SMG1 in Cholesterol Homeostasis That Depends Partially on p53 Alternative Splicing. Cancers 2022, 14, 3255. https://doi.org/10.3390/cancers14133255
Li M, Philantrope F, Diot A, Bourdon J-C, Thompson P. A Novel Role of SMG1 in Cholesterol Homeostasis That Depends Partially on p53 Alternative Splicing. Cancers. 2022; 14(13):3255. https://doi.org/10.3390/cancers14133255
Chicago/Turabian StyleLi, Muyang, Fredrick Philantrope, Alexandra Diot, Jean-Christophe Bourdon, and Patricia Thompson. 2022. "A Novel Role of SMG1 in Cholesterol Homeostasis That Depends Partially on p53 Alternative Splicing" Cancers 14, no. 13: 3255. https://doi.org/10.3390/cancers14133255
APA StyleLi, M., Philantrope, F., Diot, A., Bourdon, J.-C., & Thompson, P. (2022). A Novel Role of SMG1 in Cholesterol Homeostasis That Depends Partially on p53 Alternative Splicing. Cancers, 14(13), 3255. https://doi.org/10.3390/cancers14133255