A Genome-First Approach to Estimate Prevalence of Germline Pathogenic Variants and Risk of Pancreatic Cancer in Select Cancer Susceptibility Genes

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Exome Databases, Sequencing, and Variant Annotation
2.2. Electronic Health Records (EHR) Review
2.3. Statistical Analysis
3. Results
3.1. Frequency of GPV in Six PDAC Susceptibility Genes in All Participants and in Individuals with PDAC
3.2. Prevalence of PDAC in All Participants and in Individuals Harboring GPV in the Six Genes
3.3. Risk of PDAC in Participants Harboring GPV in ATM, BRCA1, BRCA2, CDKN2A, CHEK2, and PALB2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, G.M. Familial pancreatic cancer. Semin. Oncol. 2016, 43, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astiazaran-Symonds, E.; Goldstein, A.M. A systematic review of the prevalence of germline pathogenic variants in patients with pancreatic cancer. J. Gastroenterol. 2021, 56, 713–721. [Google Scholar] [CrossRef]
- McWilliams, R.R.; Rabe, K.G.; Olswold, C.; De Andrade, M.; Petersen, G.M. Risk of malignancy in first-degree relatives of patients with pancreatic carcinoma. Cancer 2005, 104, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, T.A. Cancer surveillance of patients from familial pancreatic cancer kindreds. Med. Clin. N. Am. 2000, 84, 707–718. [Google Scholar] [CrossRef]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef] [Green Version]
- Carey, D.J.; Fetterolf, S.N.; Davis, F.D.; Faucett, W.A.; Kirchner, H.L.; Mirshahi, U.; Murray, M.F.; Smelser, D.T.; Gerhard, G.S.; Ledbetter, D.H. The Geisinger MyCode community health initiative: An electronic health record-linked biobank for precision medicine research. Genet. Med. 2016, 18, 906–913. [Google Scholar] [CrossRef] [Green Version]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Van Hout, C.V.; Tachmazidou, I.; Backman, J.D.; Hoffman, J.D.; Liu, D.; Pandey, A.K.; Gonzaga-Jauregui, C.; Khalid, S.; Ye, B.; Banerjee, N.; et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 2020, 586, 749–756. [Google Scholar] [CrossRef]
- Backman, J.D.; Li, A.H.; Marcketta, A.; Sun, D.; Mbatchou, J.; Kessler, M.D.; Benner, C.; Liu, D.; Locke, A.E.; Balasubramanian, S.; et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 2021, 599, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivakumar, M.; Miller, J.E.; Dasari, V.R.; Gogoi, R.; Kim, D. Exome-Wide Rare Variant Analysis from the DiscovEHR Study Identifies Novel Candidate Predisposition Genes for Endometrial Cancer. Front. Oncol. 2019, 9, 574. [Google Scholar] [CrossRef] [PubMed]
- Borecka, M.; Zemankova, P.; Vocka, M.; Soucek, P.; Soukupova, J.; Kleiblova, P.; Sevcik, J.; Kleibl, Z.; Janatova, M. Mutation analysis of the PALB2 gene in unselected pancreatic cancer patients in the Czech Republic. Cancer Genet. 2016, 209, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Cremin, C.; Lee, M.K.; Hong, Q.; Hoeschen, C.; MacKenzie, A.; Dixon, K.; McCullum, M.; Nuk, J.; Kalloger, S.; Karasinska, J.; et al. Burden of hereditary cancer susceptibility in unselected patients with pancreatic ductal adenocarcinoma referred for germline screening. Cancer Med. 2020, 9, 4004–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiorzo, P.; Fornarini, G.; Sciallero, S.; Battistuzzi, L.; Belli, F.; Bernard, L.; Bonelli, L.; Borgonovo, G.; Bruno, W.; De Cian, F.; et al. CDKN2A is the main susceptibility gene in Italian pancreatic cancer families. J. Med. Genet. 2012, 49, 164–170. [Google Scholar] [CrossRef]
- Grant, R.C.; Selander, I.; Connor, A.A.; Selvarajah, S.; Borgida, A.; Briollais, L.; Petersen, G.M.; Lerner-Ellis, J.; Holter, S.; Gallinger, S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015, 148, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijers-Heijboer, H.; Ouweland, A.V.D.; Klijn, J.; Wasielewski, M.; De Snoo, A.; Oldenburg, R.; Hollestelle, A.; Houben, M.; Crepin, E.; Van Veghel-Plandsoen, M.; et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat. Genet. 2002, 31, 55–59. [Google Scholar] [PubMed]
- Hartge, P.; Struewing, J.P.; Wacholder, S.; Brody, L.C.; Tucker, M.A. The prevalence of common BRCA1 and BRCA2 mutations among Ashkenazi Jews. Am. J. Hum. Genet. 1999, 64, 963–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petridis, C.; Arora, I.; Shah, V.; Moss, C.L.; Mera, A.; Clifford, A.; Gillett, C.; Pinder, S.E.; Tomlinson, I.; Roylance, R.; et al. Frequency of Pathogenic Germline Variants in CDH1, BRCA2, CHEK2, PALB2, BRCA1, and TP53 in Sporadic Lobular Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Cavaciuti, E.; Lauge, A.; Janin, N.; Ossian, K.; Hall, J.; Stoppa-Lyonnet, D.; Andrieu, N. Cancer risk according to type and location of ATM mutation in ataxia-telangiectasia families. Genes Chromosomes Cancer 2005, 42, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, P.; Fostira, F.; Papamentzelopoulou, M.; Michelli, M.; Panopoulos, C.; Fountzilas, G.; Konstantopoulou, I.; Voutsinas, G.E.; Yannoukakos, D. CHEK2 c.1100delC allele is rarely identified in Greek breast cancer cases. Cancer Genet. 2015, 208, 129–134. [Google Scholar] [CrossRef]
- Foulkes, W.D. Inherited susceptibility to common cancers. N. Engl. J. Med. 2008, 359, 2143–2153. [Google Scholar] [CrossRef]
- Hsu, F.C.; Roberts, N.J.; Childs, E.; Porter, N.; Rabe, K.G.; Borgida, A.; Ukaegbu, C.; Goggins, M.G.; Hruban, R.H.; Zogopoulos, G.; et al. Risk of Pancreatic Cancer Among Individuals With Pathogenic Variants in the ATM Gene. JAMA Oncol. 2021, 7, 1664–1668. [Google Scholar] [CrossRef]
- Thompson, D.; Duedal, S.; Kirner, J.; McGuffog, L.; Last, J.; Reiman, A.; Byrd, P.; Taylor, M.; Easton, D.F. Cancer risks and mortality in heterozygous ATM mutation carriers. J. Natl. Cancer Inst. 2005, 97, 813–822. [Google Scholar] [CrossRef]
- Li, S.; Silvestri, V.; Leslie, G.; Rebbeck, T.R.; Neuhausen, S.L.; Hopper, J.L.; Nielsen, H.R.; Lee, A.; Yang, X.; McGuffog, L.; et al. Cancer Risks Associated With BRCA1 and BRCA2 Pathogenic Variants. J. Clin. Oncol. 2022, 40, 1529–1541. [Google Scholar] [CrossRef]
- Pilarski, R. The Role of BRCA Testing in Hereditary Pancreatic and Prostate Cancer Families. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Lener, M.R.; Kashyap, A.; Kluźniak, W.; Cybulski, C.; Soluch, A.; Pietrzak, S.; Huzarski, T.; Gronwald, J.; Lubiński, J. The Prevalence of Founder Mutations among Individuals from Families with Familial Pancreatic Cancer Syndrome. Cancer Res. Treat. 2017, 49, 430–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischkowitz, M.; Balmaña, J.; Foulkes, W.D.; James, P.; Ngeow, J.; Schmutzler, R.; Voian, N.; Wick, M.J.; Stewart, D.R.; Pal, T. Management of individuals with germline variants in PALB2: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, R.; Wang, C.; Qiu, J.; Ren, B.; You, L. Early screening and diagnosis strategies of pancreatic cancer: A comprehensive review. Cancer Commun. 2021, 41, 1257–1274. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Gordon, A.S.; Amendola, L.M.; Adelman, K.; Bale, S.J.; Chung, W.K.; Gollob, M.H.; Harrison, S.M.; Herman, G.E.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1391–1398. [Google Scholar] [CrossRef]
- Reiss, K.A.; Mick, R.; O’Hara, M.H.; Teitelbaum, U.; Karasic, T.B.; Schneider, C.; Cowden, S.; Southwell, T.; Romeo, J.; Izgur, N.; et al. Phase II Study of Maintenance Rucaparib in Patients with Platinum-Sensitive Advanced Pancreatic Cancer and a Pathogenic Germline or Somatic Variant in BRCA1, BRCA2, or PALB2. J. Clin. Oncol. 2021, 39, 2497–2505. [Google Scholar] [CrossRef]
- Truty, R.; Paul, J.; Kennemer, M.; Lincoln, S.E.; Olivares, E.; Nussbaum, R.L.; Aradhya, S. Prevalence and properties of intragenic copy-number variation in Mendelian disease genes. Genet. Med. 2019, 21, 114–123. [Google Scholar] [CrossRef] [Green Version]
- LaDuca, H.; Polley, E.C.; Yussuf, A.; Hoang, L.; Bs, S.G.; Hart, S.N.; Yadav, S.; Hu, C.; Na Ms, J.; Goldgar, D.E.; et al. A clinical guide to hereditary cancer panel testing: Evaluation of gene-specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high-risk patients. Genet. Med. 2020, 22, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, A.M.; Chan, M.; Harland, M.; Gillanders, E.M.; Hayward, N.K.; Avril, M.-F.; Azizi, E.; Bianchi-Scarra, G.; Bishop, D.T.; Paillerets, B.B.-D.; et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006, 66, 9818–9828. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
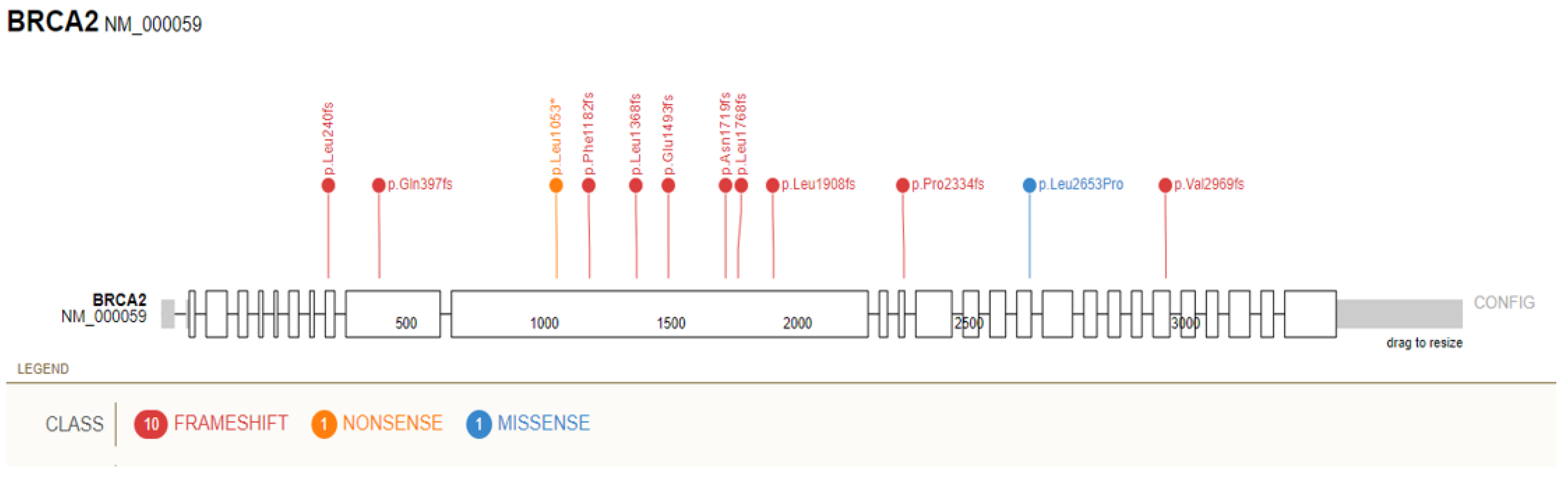{kind=link}
{kind=link}
| Number and Prevalence (%, in Parentheses) of Heterozygotes for GPV in UKB (n = 200,619) | Number and Prevalence (%, in Parentheses) of Heterozygotes for GPV in GHS (n = 175,449) | Number and Prevalence (%, in Parentheses) of Heterozygotes for GPV in Both Cohorts (n = 376,068) | |
|---|---|---|---|
| ATM | 839 (0.42%) | 885 (0.50%) | 1724 (0.46%) |
| BRCA1 | 223 (0.11%) | 356 (0.20%) | 579 (0.15%) |
| BRCA2 | 710 (0.35%) | 717 (0.40%) | 1427 (0.38%) |
| CDKN2A | 137 (0.07%) | 47 (0.027%) | 184 (0.05%) |
| CHEK2 | 1619 (0.80%) | 2987 (1.69%) | 4606 (1.22%) |
| PALB2 | 397 (0.20%) | 205 (0.12%) | 6022 (0.16%) |
| UKB (n = 417) | GHS (n = 592) | Both Cohorts (n = 1009) | Astiazaran-Symonds et al., 2021 | |||
|---|---|---|---|---|---|---|
| Number of PDAC Participants with GPVs | Frequency of GPVs in PDAC Participants | Number of PDAC Participants with GPVs | Frequency of GPVs in PDAC Participants | Cumulative Frequency | Cumulative Frequency | |
| ALL P/LP | 25 * | 5.99% | 45 | 7.60% | 6.94% | - |
| ATM | 11 | 2.64% | 13 | 2.19% | 2.38% | 2.52% |
| BRCA1 | 0 | 0.0% | 2 | 0.34% | 0.19% | 0.99% |
| BRCA2 | 5 | 1.20% | 9 | 1.52% | 1.38% | 2.90% |
| CDKN2A | 2 | 0.48% | 0 | 0.0% | 0.19% | 0.98% |
| CHEK2 | 6 | 1.44% | 16 | 2.70% | 2.18% | 1.15% |
| PALB2 | 2 | 0.48% | 5 | 0.84% | 0.69% | 0.65% |
| UKB Participants Harboring GPVs Who Developed PDAC | GHS Participants Harboring GPVs Who Developed PDAC | Total Number of Heterozygotes for GPVs Who Developed PDAC | |
|---|---|---|---|
| ATM | 11/839 (1.31%) | 13/885 (1.47%) | 24/1724 (1.39%) |
| BRCA1 | 0/223 (0%) | 2/356 (0.56%) | 2/579 (0.34%) |
| BRCA2 | 5/710 (0.70%) | 9/717 (1.25%) | 14/1427 (0.98%) |
| CDKN2A | 2/137 (1.45%) | 0/47 (0%) | 2/184 (1.09%) |
| CHEK2 | 6/1619 (0.37%) | 16/2987 (0.53%) | 22/4606 (0.48%) |
| PALB2 | 2/397 (0.50%) | 5/205 (2.44%) | 7/602 (1.16%) |
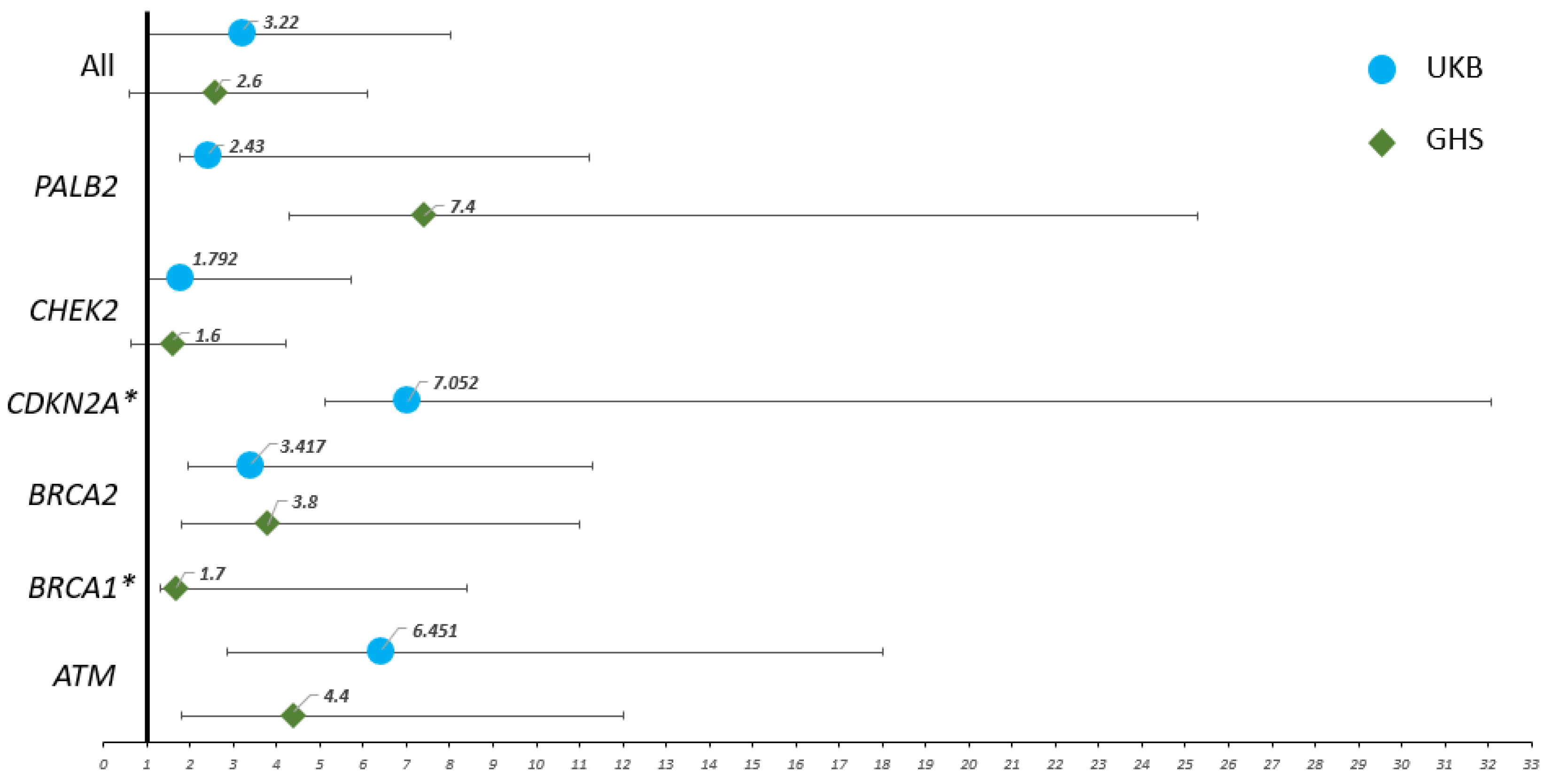
| UKB | GHS | |||
|---|---|---|---|---|
| Gene | RR (95% CI | p-Value | RR | p-Value |
| ATM | 6.4 (3.6–11.6) | <0.0001 * | 4.4 (2.6–7.6) | <0.0001 * |
| BRCA1 | N/A | >0.9999 | 1.7 (0.4–6.7) | 0.3176 |
| BRCA2 | 3.4 (1.4–7.9) | 0.0151 * | 3.8 (2–7.2) | 0.0007 * |
| CDKN2A | 7 (1.9–25) | 0.0312 * | N/A | >0.9999 |
| CHEK2 | 1.8 (0.8–3.9) | 0.1482 | 1.6 (1–2.6) | 0.0411 * |
| PALB2 | 2.4 (0.7–8.8) | 0.1888 | 7.4 (3.1–17.9) | 0.0006 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Astiazaran-Symonds, E.; Kim, J.; Haley, J.S.; Kim, S.Y.; Rao, H.S.; Genetics Center, R.; Carey, D.J.; Stewart, D.R.; Goldstein, A.M. A Genome-First Approach to Estimate Prevalence of Germline Pathogenic Variants and Risk of Pancreatic Cancer in Select Cancer Susceptibility Genes. Cancers 2022, 14, 3257. https://doi.org/10.3390/cancers14133257
Astiazaran-Symonds E, Kim J, Haley JS, Kim SY, Rao HS, Genetics Center R, Carey DJ, Stewart DR, Goldstein AM. A Genome-First Approach to Estimate Prevalence of Germline Pathogenic Variants and Risk of Pancreatic Cancer in Select Cancer Susceptibility Genes. Cancers. 2022; 14(13):3257. https://doi.org/10.3390/cancers14133257
Chicago/Turabian StyleAstiazaran-Symonds, Esteban, Jung Kim, Jeremy S. Haley, Sun Young Kim, H. Shanker Rao, Regeneron Genetics Center, David J. Carey, Douglas R. Stewart, and Alisa M. Goldstein. 2022. "A Genome-First Approach to Estimate Prevalence of Germline Pathogenic Variants and Risk of Pancreatic Cancer in Select Cancer Susceptibility Genes" Cancers 14, no. 13: 3257. https://doi.org/10.3390/cancers14133257
APA StyleAstiazaran-Symonds, E., Kim, J., Haley, J. S., Kim, S. Y., Rao, H. S., Genetics Center, R., Carey, D. J., Stewart, D. R., & Goldstein, A. M. (2022). A Genome-First Approach to Estimate Prevalence of Germline Pathogenic Variants and Risk of Pancreatic Cancer in Select Cancer Susceptibility Genes. Cancers, 14(13), 3257. https://doi.org/10.3390/cancers14133257