
The Desmoplastic Stroma of Pancreatic Cancer: Multilayered Levels of Heterogeneity, Clinical Significance, and Therapeutic Opportunities


Abstract
:Simple Summary
Abstract
1. Introduction
2. Extracellular Matrix
2.1. Collagens
2.2. Hyaluronan
2.3. Laminins
3. Fibroblast Heterogeneity
3.1. CAF Origins
3.2. CAF Markers
3.3. CAF Subtypes
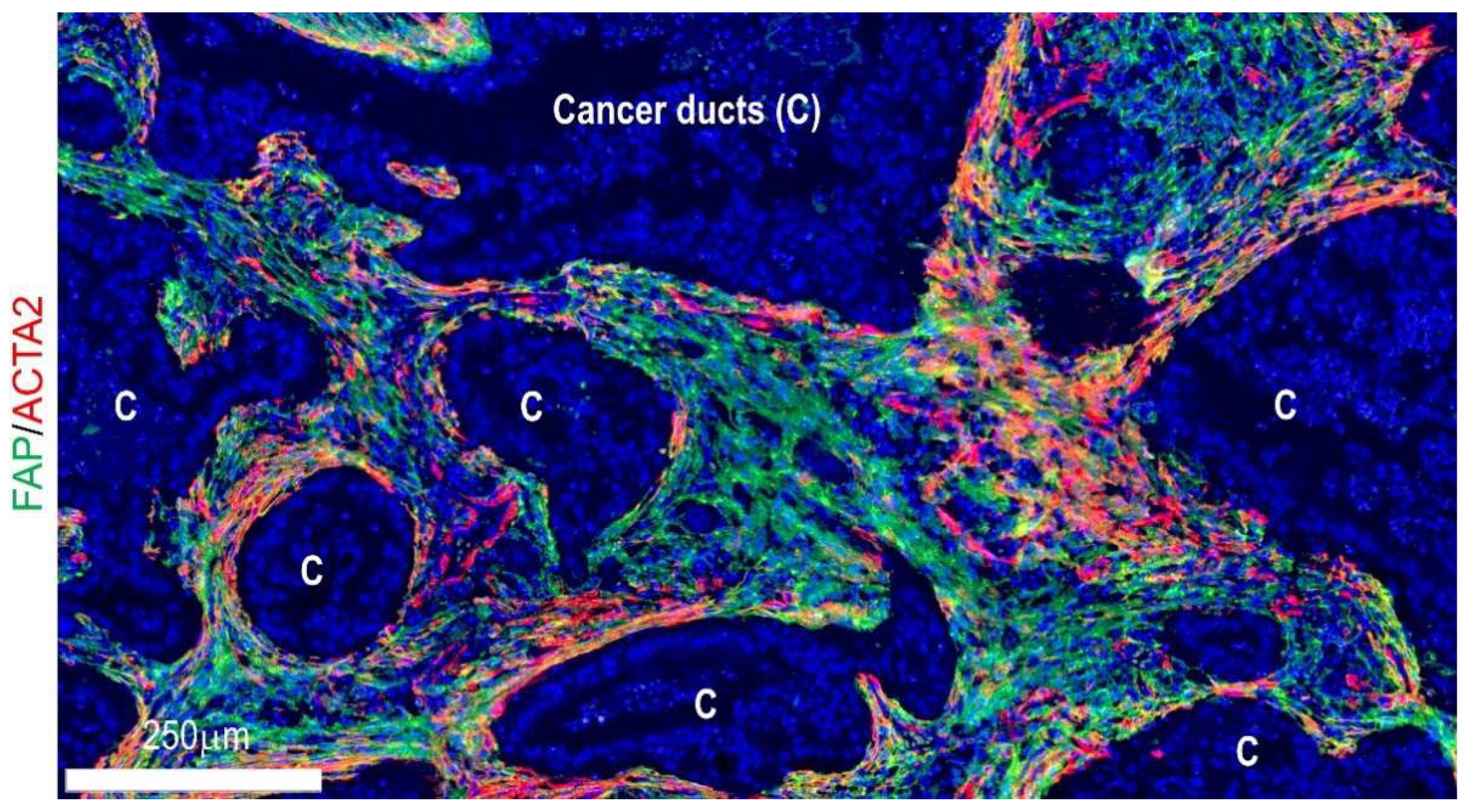
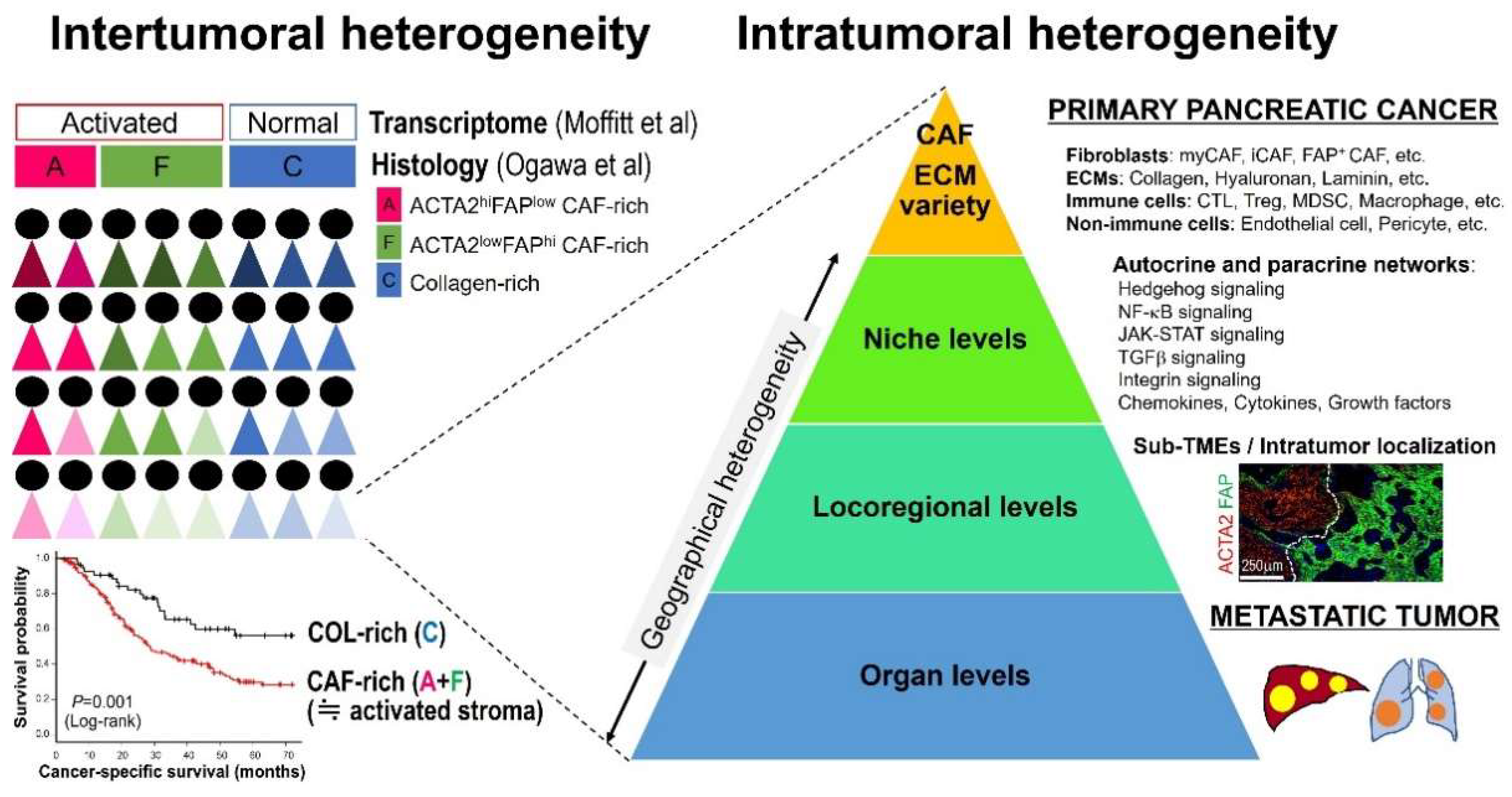
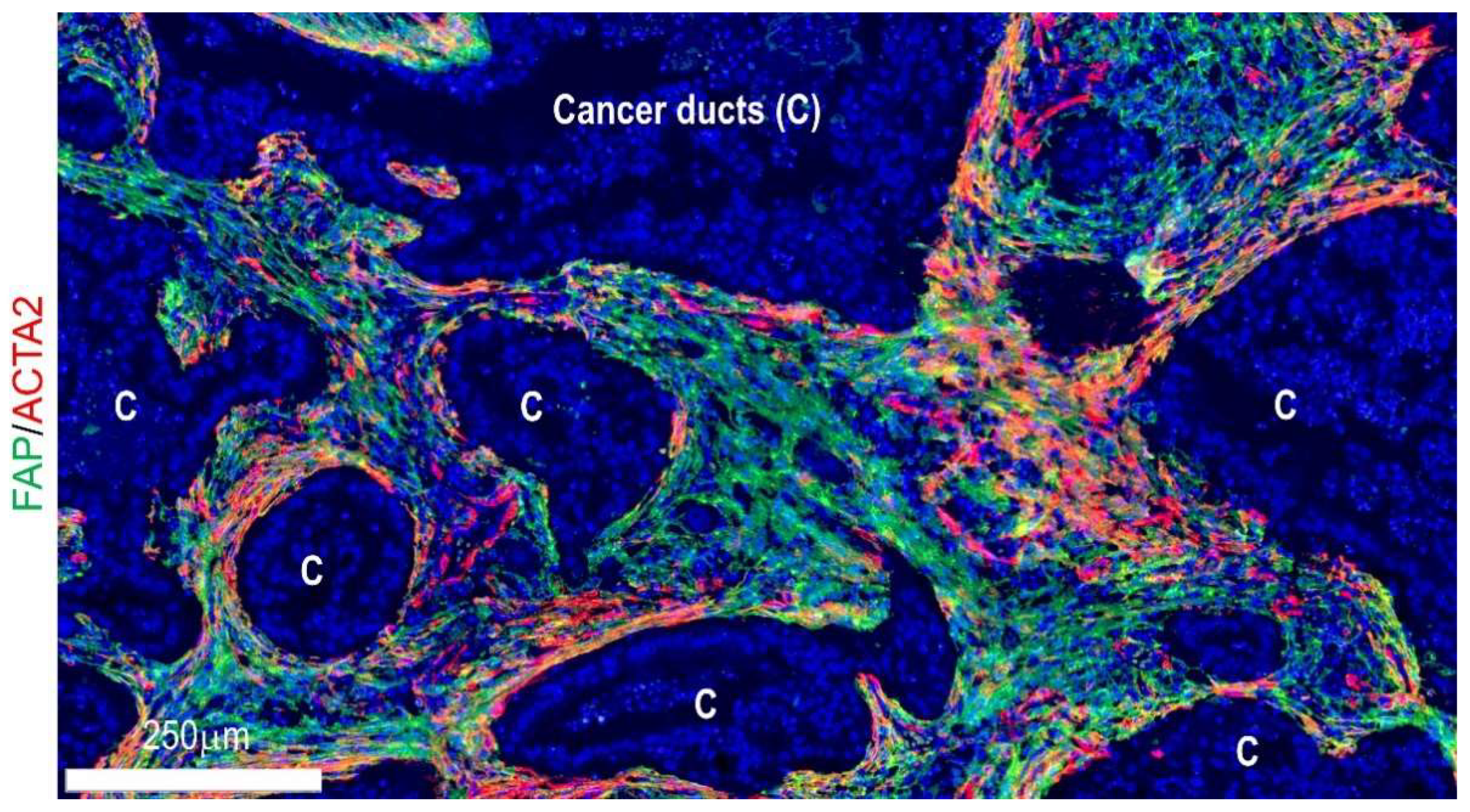
4. Geographical Heterogeneity
4.1. Cell–Cell Interactions and Signaling at Niche Levels
4.2. Locoregional Levels
4.3. Organ Levels
5. Intertumoral Stromal Heterogeneity and Clinical Implications
6. Future Direction of Targeting Desmoplastic Stroma of Pancreatic Cancer
7. Conclusions
- Use of Standardized Official Symbols
Funding
Conflicts of Interest
References
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Klein, A.P. Pancreatic cancer: A growing burden. Lancet Gastroenterol. Hepatol. 2019, 4, 895–896. [Google Scholar] [CrossRef] [Green Version]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Hahn, S.A.; Real, F.X. A genetic roadmap of pancreatic cancer: Still evolving. Gut 2017, 66, 2170–2178. [Google Scholar] [CrossRef]
- Hayashi, A.; Hong, J.; Iacobuzio-Donahue, C.A. The pancreatic cancer genome revisited. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 469–481. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, B.; Li, H. The Roles of Frequently Mutated Genes of Pancreatic Cancer in Regulation of Tumor Microenvironment. Technol. Cancer Res. Treat. 2020, 19, 1533033820920969. [Google Scholar] [CrossRef]
- Bazzichetto, C.; Luchini, C.; Conciatori, F.; Vaccaro, V.; Di Cello, I.; Mattiolo, P.; Falcone, I.; Ferretti, G.; Scarpa, A.; Cognetti, F.; et al. Morphologic and Molecular Landscape of Pancreatic Cancer Variants as the Basis of New Therapeutic Strategies for Precision Oncology. Int. J. Mol. Sci. 2020, 21, 8841. [Google Scholar] [CrossRef]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef]
- Ren, B.; Liu, X.; Suriawinata, A.A. Pancreatic Ductal Adenocarcinoma and Its Precursor Lesions: Histopathology, Cytopathology, and Molecular Pathology. Am. J. Pathol. 2019, 189, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Parkin, A.; Man, J.; Chou, A.; Nagrial, A.M.; Samra, J.; Gill, A.J.; Timpson, P.; Pajic, M. The Evolving Understanding of the Molecular and Therapeutic Landscape of Pancreatic Ductal Adenocarcinoma. Diseases 2018, 6, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, K.; Masugi, Y.; Effendi, K.; Tsujikawa, H.; Hiraoka, N.; Kitago, M.; Shinoda, M.; Itano, O.; Tanabe, M.; Kitagawa, Y.; et al. Upregulated SMAD3 promotes epithelial-mesenchymal transition and predicts poor prognosis in pancreatic ductal adenocarcinoma. Lab. Investig. 2014, 94, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.R.; Rubinson, D.A.; Nowak, J.A.; Morales-Oyarvide, V.; Dunne, R.F.; Kozak, M.M.; Welch, M.W.; Brais, L.K.; Da Silva, A.; Li, T.; et al. Association of Alterations in Main Driver Genes with Outcomes of Patients With Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2018, 4, e173420. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Nowak, J.A.; Camarda, N.D.; Moffitt, R.A.; Ghazani, A.A.; Hazar-Rethinam, M.; Raghavan, S.; Kim, J.; Brais, L.K.; Ragon, D.; et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018, 8, 1096–1111. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.D.; Roberts, N.J.; Wood, L.D.; Eshleman, J.R.; Goggins, M.G.; Kern, S.E.; Klein, A.P.; Hruban, R.H. The genetics of ductal adenocarcinoma of the pancreas in the year 2020: Dramatic progress, but far to go. Mod. Pathol. 2020, 33, 2544–2563. [Google Scholar] [CrossRef]
- Connor, A.A.; Gallinger, S. Next generation sequencing of pancreatic ductal adenocarcinoma: Right or wrong? Expert Rev. Gastroenterol. Hepatol. 2017, 11, 683–694. [Google Scholar] [CrossRef]
- Pihlak, R.; Weaver, J.M.J.; Valle, J.W.; McNamara, M.G. Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy. Cancers 2018, 10, 17. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e1993. [Google Scholar] [CrossRef] [Green Version]
- Fujita, Y.; Matsuda, S.; Sasaki, Y.; Masugi, Y.; Kitago, M.; Yagi, H.; Abe, Y.; Shinoda, M.; Tokino, T.; Sakamoto, M.; et al. Pathogenesis of multiple pancreatic cancers involves multicentric carcinogenesis and intrapancreatic metastasis. Cancer Sci. 2020, 111, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Luchini, C.; Grant, R.C.; Scarpa, A.; Gallinger, S. Microsatellite instability/mismatch repair deficiency in pancreatic cancers: The same or different? Gut 2021, 70, 1809–1811. [Google Scholar] [CrossRef]
- Perkhofer, L.; Gout, J.; Roger, E.; Kude de Almeida, F.; Baptista Simoes, C.; Wiesmuller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2021, 70, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; O’Kane, G.M.; Denroche, R.E.; Raitses-Gurevich, M.; Grant, R.C.; Holter, S.; Wang, Y.; Zhang, A.; Jang, G.H.; Stossel, C.; et al. Genomic Features and Classification of Homologous Recombination Deficient Pancreatic Ductal Adenocarcinoma. Gastroenterology 2021, 160, 2119–2132.e2119. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, V.; Leslie, G.; Barnes, D.R.; Group, C.; Agnarsson, B.A.; Aittomaki, K.; Alducci, E.; Andrulis, I.L.; Barkardottir, R.B.; Barroso, A.; et al. Characterization of the Cancer Spectrum in Men with Germline BRCA1 and BRCA2 Pathogenic Variants: Results From the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). JAMA Oncol. 2020, 6, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e2244. [Google Scholar] [CrossRef] [Green Version]
- Hester, R.; Mazur, P.K.; McAllister, F. Immunotherapy in Pancreatic Adenocarcinoma: Beyond “Copy/Paste”. Clin. Cancer Res. 2021, 27, 6287–6297. [Google Scholar] [CrossRef]
- Neesse, A.; Bauer, C.A.; Ohlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2019, 68, 159–171. [Google Scholar] [CrossRef]
- Bhagat, T.D.; Von Ahrens, D.; Dawlaty, M.; Zou, Y.; Baddour, J.; Achreja, A.; Zhao, H.; Yang, L.; Patel, B.; Kwak, C.; et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. eLife 2019, 8, e50663. [Google Scholar] [CrossRef]
- Bahrami, A.; Khazaei, M.; Bagherieh, F.; Ghayour-Mobarhan, M.; Maftouh, M.; Hassanian, S.M.; Avan, A. Targeting stroma in pancreatic cancer: Promises and failures of targeted therapies. J. Cell. Physiol. 2017, 232, 2931–2937. [Google Scholar] [CrossRef]
- Suzuki, T.; Masugi, Y.; Inoue, Y.; Hamada, T.; Tanaka, M.; Takamatsu, M.; Arita, J.; Kato, T.; Kawaguchi, Y.; Kunita, A.; et al. KRAS variant allele frequency, but not mutation positivity, associates with survival of patients with pancreatic cancer. Cancer Sci. 2022; early view. [Google Scholar] [CrossRef]
- Ferrara, B.; Pignatelli, C.; Cossutta, M.; Citro, A.; Courty, J.; Piemonti, L. The Extracellular Matrix in Pancreatic Cancer: Description of a Complex Network and Promising Therapeutic Options. Cancers 2021, 13, 4442. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Storz, P.; Crawford, H.C. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2020, 158, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. A centralized communication network: Recent insights into the role of the cancer associated fibroblast in the development of drug resistance in tumors. Semin. Cell Dev. Biol. 2020, 101, 111–114. [Google Scholar] [CrossRef]
- Morita, T.; Kodama, Y.; Shiokawa, M.; Kuriyama, K.; Marui, S.; Kuwada, T.; Sogabe, Y.; Matsumori, T.; Kakiuchi, N.; Tomono, T.; et al. CXCR4 in Tumor Epithelial Cells Mediates Desmoplastic Reaction in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2020, 80, 4058–4070. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 5, e129212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e127. [Google Scholar] [CrossRef] [PubMed]
- Miyabayashi, K.; Baker, L.A.; Deschenes, A.; Traub, B.; Caligiuri, G.; Plenker, D.; Alagesan, B.; Belleau, P.; Li, S.; Kendall, J.; et al. Intraductal Transplantation Models of Human Pancreatic Ductal Adenocarcinoma Reveal Progressive Transition of Molecular Subtypes. Cancer Discov. 2020, 10, 1566–1589. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Masugi, Y.; Abe, T.; Yamazaki, K.; Ueno, A.; Fujii-Nishimura, Y.; Hori, S.; Yagi, H.; Abe, Y.; Kitago, M.; et al. Three Distinct Stroma Types in Human Pancreatic Cancer Identified by Image Analysis of Fibroblast Subpopulations and Collagen. Clin. Cancer Res. 2021, 27, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Clauser, K.R.; Ohlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, R.; Luong, T.; Cao, X.; Han, B.; Shah, N.; Franco-Barraza, J.; Han, L.; Shenoy, V.B.; Lelkes, P.I.; Cukierman, E. Rigidity controls human desmoplastic matrix anisotropy to enable pancreatic cancer cell spread via extracellular signal-regulated kinase 2. Matrix Biol. 2019, 81, 50–69. [Google Scholar] [CrossRef]
- Nieskoski, M.D.; Marra, K.; Gunn, J.R.; Hoopes, P.J.; Doyley, M.M.; Hasan, T.; Trembly, B.S.; Pogue, B.W. Collagen Complexity Spatially Defines Microregions of Total Tissue Pressure in Pancreatic Cancer. Sci. Rep. 2017, 7, 10093. [Google Scholar] [CrossRef]
- Nia, H.T.; Munn, L.L.; Jain, R.K. Physical traits of cancer. Science 2020, 370, eaaz0868. [Google Scholar] [CrossRef]
- Maneshi, P.; Mason, J.; Dongre, M.; Ohlund, D. Targeting Tumor-Stromal Interactions in Pancreatic Cancer: Impact of Collagens and Mechanical Traits. Front. Cell Dev. Biol. 2021, 9, 787485. [Google Scholar] [CrossRef]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Taipale, J.; Keski-Oja, J. Growth factors in the extracellular matrix. FASEB J. 1997, 11, 51–59. [Google Scholar] [CrossRef]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cells: Aiding and abetting pancreatic cancer progression. Pancreatology 2020, 20, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.C.; Fu, J.C.; Uto, N.; Horiuchi, K.; Fujimoto, D. Collagen cross-linking: Lysyl oxidase dependent synthesis of pyridinoline in vitro: Confirmation that pyridinoline is derived from collagen. Biochem. Biophys. Res. Commun. 1982, 108, 1546–1550. [Google Scholar] [CrossRef]
- Setargew, Y.F.I.; Wyllie, K.; Grant, R.D.; Chitty, J.L.; Cox, T.R. Targeting Lysyl Oxidase Family Meditated Matrix Cross-Linking as an Anti-Stromal Therapy in Solid Tumours. Cancers 2021, 13, 491. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Morton, J.P. LOXL2 in pancreatic tumourigenesis: The complexity of tumour-stromal crosstalk exemplified. Gut 2022. [Google Scholar] [CrossRef]
- Alonso-Nocelo, M.; Ruiz-Canas, L.; Sancho, P.; Gorgulu, K.; Alcala, S.; Pedrero, C.; Vallespinos, M.; Lopez-Gil, J.C.; Ochando, M.; Garcia-Garcia, E.; et al. Macrophages direct cancer cells through a LOXL2-mediated metastatic cascade in pancreatic ductal adenocarcinoma. Gut 2022. [Google Scholar] [CrossRef]
- Benson, A.B., 3rd; Wainberg, Z.A.; Hecht, J.R.; Vyushkov, D.; Dong, H.; Bendell, J.; Kudrik, F. A Phase II Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination with Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma. Oncologist 2017, 22, 241.e215. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy with FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Lockhart, A.C.; Tan, B.R.; Suresh, R.; Lim, K.-H.; Ratner, L.; Morton, A.; Huffman, J.; Marquez, S.; Boice, N.; et al. Phase I study of defactinib combined with pembrolizumab and gemcitabine in patients with advanced cancer. J. Clin. Oncol. 2018, 36, 380. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients with Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients with Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M.A.; Cutsem, E.V.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. HALO 109-301: A randomized, double-blind, placebo-controlled, phase 3 study of pegvorhyaluronidase alfa (PEGPH20) + nab-paclitaxel/gemcitabine (AG) in patients (pts) with previously untreated hyaluronan (HA)-high metastatic pancreatic ductal adenocarcinoma (mPDA). J. Clin. Oncol. 2020, 38, 638. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Bockorny, B.; Macarulla, T.; Semenisty, V.; Borazanci, E.; Feliu, J.; Ponz-Sarvise, M.; Abad, D.G.; Oberstein, P.; Alistar, A.; Munoz, A.; et al. Motixafortide and Pembrolizumab Combined to Nanoliposomal Irinotecan, Fluorouracil, and Folinic Acid in Metastatic Pancreatic Cancer: The COMBAT/KEYNOTE-202 Trial. Clin. Cancer Res. 2021, 27, 5020–5027. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Vasvani, S.; Kulkarni, P.; Rawtani, D. Hyaluronic acid: A review on its biology, aspects of drug delivery, route of administrations and a special emphasis on its approved marketed products and recent clinical studies. Int. J. Biol. Macromol. 2020, 151, 1012–1029. [Google Scholar] [CrossRef] [PubMed]
- Zadnikova, P.; Sinova, R.; Pavlik, V.; Simek, M.; Safrankova, B.; Hermannova, M.; Nesporova, K.; Velebny, V. The Degradation of Hyaluronan in the Skin. Biomolecules 2022, 12, 251. [Google Scholar] [CrossRef] [PubMed]
- Jegasothy, S.M.; Zabolotniaia, V.; Bielfeldt, S. Efficacy of a New Topical Nano-hyaluronic Acid in Humans. J. Clin. Aesthet. Dermatol. 2014, 7, 27–29. [Google Scholar]
- Valle, S.; Martin-Hijano, L.; Alcala, S.; Alonso-Nocelo, M.; Sainz, B., Jr. The Ever-Evolving Concept of the Cancer Stem Cell in Pancreatic Cancer. Cancers 2018, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Blair, A.B.; Kim, V.M.; Muth, S.T.; Saung, M.T.; Lokker, N.; Blouw, B.; Armstrong, T.D.; Jaffee, E.M.; Tsujikawa, T.; Coussens, L.M.; et al. Dissecting the Stromal Signaling and Regulation of Myeloid Cells and Memory Effector T Cells in Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 5351–5363. [Google Scholar] [CrossRef]
- Li, X.; Shepard, H.M.; Cowell, J.A.; Zhao, C.; Osgood, R.J.; Rosengren, S.; Blouw, B.; Garrovillo, S.A.; Pagel, M.D.; Whatcott, C.J.; et al. Parallel Accumulation of Tumor Hyaluronan, Collagen, and Other Drivers of Tumor Progression. Clin. Cancer Res. 2018, 24, 4798–4807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, H.; Luo, J.; Wu, X.; Li, X.; Zhao, X.; Zhou, D.; Yu, S. Overexpression of alpha3, beta3 and gamma2 chains of laminin-332 is associated with poor prognosis in pancreatic ductal adenocarcinoma. Oncol. Lett. 2018, 16, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Masugi, Y.; Yamazaki, K.; Emoto, K.; Effendi, K.; Tsujikawa, H.; Kitago, M.; Itano, O.; Kitagawa, Y.; Sakamoto, M. Upregulation of integrin beta4 promotes epithelial-mesenchymal transition and is a novel prognostic marker in pancreatic ductal adenocarcinoma. Lab. Investig. 2015, 95, 308–319. [Google Scholar] [CrossRef] [Green Version]
- Tani, T.; Lumme, A.; Linnala, A.; Kivilaakso, E.; Kiviluoto, T.; Burgeson, R.E.; Kangas, L.; Leivo, I.; Virtanen, I. Pancreatic carcinomas deposit laminin-5, preferably adhere to laminin-5, and migrate on the newly deposited basement membrane. Am. J. Pathol. 1997, 151, 1289–1302. [Google Scholar]
- Guo, W.; Pylayeva, Y.; Pepe, A.; Yoshioka, T.; Muller, W.J.; Inghirami, G.; Giancotti, F.G. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 2006, 126, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, A.M.; Rabinovitz, I. Towards a mechanistic understanding of tumor invasion—lessons from the alpha6beta 4 integrin. Semin. Cancer Biol. 2001, 11, 129–141. [Google Scholar] [CrossRef]
- Okada, Y.; Takahashi, N.; Takayama, T.; Goel, A. LAMC2 promotes cancer progression and gemcitabine resistance through modulation of EMT and ATP-binding cassette transporters in pancreatic ductal adenocarcinoma. Carcinogenesis 2021, 42, 546–556. [Google Scholar] [CrossRef]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Liu Donaher, J.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef] [Green Version]
- Ruan, S.; Lin, M.; Zhu, Y.; Lum, L.; Thakur, A.; Jin, R.; Shao, W.; Zhang, Y.; Hu, Y.; Huang, S.; et al. Integrin beta4-Targeted Cancer Immunotherapies Inhibit Tumor Growth and Decrease Metastasis. Cancer Res. 2020, 80, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Giancotti, F.G. Targeting integrin beta4 for cancer and anti-angiogenic therapy. Trends Pharmacol. Sci. 2007, 28, 506–511. [Google Scholar] [CrossRef]
- Dobson, H.E.; Ruan, S.; Chang, A.E.; Wicha, M.S.; Li, Q. Targeting cancer stem cells via integrin beta4. Oncotarget 2021, 12, 1850–1858. [Google Scholar] [CrossRef]
- Barrera, L.N.; Evans, A.; Lane, B.; Brumskill, S.; Oldfield, F.E.; Campbell, F.; Andrews, T.; Lu, Z.; Perez-Mancera, P.A.; Liloglou, T.; et al. Fibroblasts from Distinct Pancreatic Pathologies Exhibit Disease-Specific Properties. Cancer Res. 2020, 80, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Lafaro, K.J.; Melstrom, L.G. The Paradoxical Web of Pancreatic Cancer Tumor Microenvironment. Am. J. Pathol. 2019, 189, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Cannon, A.; Thompson, C.; Hall, B.R.; Jain, M.; Kumar, S.; Batra, S.K. Desmoplasia in pancreatic ductal adenocarcinoma: Insight into pathological function and therapeutic potential. Genes Cancer 2018, 9, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Cannon, A.; Thompson, C.M.; Bhatia, R.; Armstrong, K.A.; Solheim, J.C.; Kumar, S.; Batra, S.K. Molecular mechanisms of pancreatic myofibroblast activation in chronic pancreatitis and pancreatic ductal adenocarcinoma. J. Gastroenterol. 2021, 56, 689–703. [Google Scholar] [CrossRef]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Whittle, M.C.; Hingorani, S.R. Fibroblasts in Pancreatic Ductal Adenocarcinoma: Biological Mechanisms and Therapeutic Targets. Gastroenterology 2019, 156, 2085–2096. [Google Scholar] [CrossRef]
- Sano, M.; Driscoll, D.R.; De Jesus-Monge, W.E.; Klimstra, D.S.; Lewis, B.C. Activated wnt signaling in stroma contributes to development of pancreatic mucinous cystic neoplasms. Gastroenterology 2014, 146, 257–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ideno, N.; Yamaguchi, H.; Ghosh, B.; Gupta, S.; Okumura, T.; Steffen, D.J.; Fisher, C.G.; Wood, L.D.; Singhi, A.D.; Nakamura, M.; et al. GNAS(R201C) Induces Pancreatic Cystic Neoplasms in Mice That Express Activated KRAS by Inhibiting YAP1 Signaling. Gastroenterology 2018, 155, 1593–1607.e1512. [Google Scholar] [CrossRef] [PubMed]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.X.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A.; et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007, 11, 229–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taki, K.; Ohmuraya, M.; Tanji, E.; Komatsu, H.; Hashimoto, D.; Semba, K.; Araki, K.; Kawaguchi, Y.; Baba, H.; Furukawa, T. GNAS(R201H) and Kras(G12D) cooperate to promote murine pancreatic tumorigenesis recapitulating human intraductal papillary mucinous neoplasm. Oncogene 2016, 35, 2407–2412. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Bernard, V.; Semaan, A.; Huang, J.; San Lucas, F.A.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res. 2019, 25, 2194–2205. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.E.; Adoumie, M.; Kim, E.C.; Zhang, Y.; Scales, M.K.; El-Tawil, Y.S.; Shaikh, A.Z.; Wen, H.J.; Bednar, F.; Allen, B.L.; et al. Differential Contribution of Pancreatic Fibroblast Subsets to the Pancreatic Cancer Stroma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 581–599. [Google Scholar] [CrossRef]
- Zhu, K.; Cai, L.; Cui, C.; de Los Toyos, J.R.; Anastassiou, D. Single-cell analysis reveals the pan-cancer invasiveness-associated transition of adipose-derived stromal cells into COL11A1-expressing cancer-associated fibroblasts. PLoS Comput. Biol. 2021, 17, e1009228. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef]
- Huang, H.; Wang, Z.; Zhang, Y.; Pradhan, R.N.; Ganguly, D.; Chandra, R.; Murimwa, G.; Wright, S.; Gu, X.; Maddipati, R.; et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 2022, 40, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Vaish, U.; Jain, T.; Are, A.C.; Dudeja, V. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting. Int. J. Mol. Sci. 2021, 22, 13408. [Google Scholar] [CrossRef]
- Manoukian, P.; Bijlsma, M.; van Laarhoven, H. The Cellular Origins of Cancer-Associated Fibroblasts and Their Opposing Contributions to Pancreatic Cancer Growth. Front. Cell. Dev. Biol. 2021, 9, 743907. [Google Scholar] [CrossRef]
- Helms, E.J.; Berry, M.W.; Chaw, R.C.; DuFort, C.C.; Sun, D.; Onate, M.K.; Oon, C.; Bhattacharyya, S.; Sanford-Crane, H.; Horton, W.; et al. Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer-Associated Fibroblasts. Cancer Discov. 2022, 12, 484–501. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Wang, L.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [Green Version]
- Cremasco, V.; Astarita, J.L.; Grauel, A.L.; Keerthivasan, S.; MacIsaac, K.; Woodruff, M.C.; Wu, M.; Spel, L.; Santoro, S.; Amoozgar, Z.; et al. FAP Delineates Heterogeneous and Functionally Divergent Stromal Cells in Immune-Excluded Breast Tumors. Cancer Immunol. Res. 2018, 6, 1472–1485. [Google Scholar] [CrossRef] [Green Version]
- Awaji, M.; Singh, R.K. Cancer-Associated Fibroblasts’ Functional Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancers 2019, 11, 290. [Google Scholar] [CrossRef] [Green Version]
- Galbo, P.M., Jr.; Zang, X.; Zheng, D. Molecular Features of Cancer-associated Fibroblast Subtypes and their Implication on Cancer Pathogenesis, Prognosis, and Immunotherapy Resistance. Clin. Cancer Res. 2021, 27, 2636–2647. [Google Scholar] [CrossRef]
- Hu, B.; Wu, C.; Mao, H.; Gu, H.; Dong, H.; Yan, J.; Qi, Z.; Yuan, L.; Dong, Q.; Long, J. Subpopulations of cancer-associated fibroblasts link the prognosis and metabolic features of pancreatic ductal adenocarcinoma. Ann. Transl. Med. 2022, 10, 262. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Q.; Li, M.; Guo, H.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine 2021, 66, 103315. [Google Scholar] [CrossRef] [PubMed]
- Crawford, H.C.; Pasca di Magliano, M.; Banerjee, S. Signaling Networks That Control Cellular Plasticity in Pancreatic Tumorigenesis, Progression, and Metastasis. Gastroenterology 2019, 156, 2073–2084. [Google Scholar] [CrossRef] [PubMed]
- Uzunparmak, B.; Sahin, I.H. Pancreatic cancer microenvironment: A current dilemma. Clin. Transl. Med. 2019, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Vennin, C.; Melenec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Hutton, C.; Heider, F.; Blanco-Gomez, A.; Banyard, A.; Kononov, A.; Zhang, X.; Karim, S.; Paulus-Hock, V.; Watt, D.; Steele, N.; et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 2021, 39, 1227–1244.e1220. [Google Scholar] [CrossRef]
- Francescone, R.; Barbosa Vendramini-Costa, D.; Franco-Barraza, J.; Wagner, J.; Muir, A.; Lau, A.N.; Gabitova, L.; Pazina, T.; Gupta, S.; Luong, T.; et al. Netrin G1 Promotes Pancreatic Tumorigenesis through Cancer-Associated Fibroblast-Driven Nutritional Support and Immunosuppression. Cancer Discov. 2021, 11, 446–479. [Google Scholar] [CrossRef]
- Awaji, M.; Saxena, S.; Wu, L.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M.; et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 2020, 34, 9405–9418. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; DeNardo, D.G. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol. Immunother. CII 2017, 66, 1037–1048. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol. Cancer 2018, 17, 62. [Google Scholar] [CrossRef] [Green Version]
- Vennin, C.; Chin, V.T.; Warren, S.C.; Lucas, M.C.; Herrmann, D.; Magenau, A.; Melenec, P.; Walters, S.N.; Del Monte-Nieto, G.; Conway, J.R.; et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci. Transl. Med. 2017, 9, eaai8504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Parente, P.; Parcesepe, P.; Covelli, C.; Olivieri, N.; Remo, A.; Pancione, M.; Latiano, T.P.; Graziano, P.; Maiello, E.; Giordano, G. Crosstalk between the Tumor Microenvironment and Immune System in Pancreatic Ductal Adenocarcinoma: Potential Targets for New Therapeutic Approaches. Gastroenterol. Res. Pract. 2018, 2018, 7530619. [Google Scholar] [CrossRef] [PubMed]
- Elahi-Gedwillo, K.Y.; Carlson, M.; Zettervall, J.; Provenzano, P.P. Antifibrotic Therapy Disrupts Stromal Barriers and Modulates the Immune Landscape in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 372–386. [Google Scholar] [CrossRef] [Green Version]
- Masugi, Y.; Abe, T.; Ueno, A.; Fujii-Nishimura, Y.; Ojima, H.; Endo, Y.; Fujita, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; et al. Characterization of spatial distribution of tumor-infiltrating CD8(+) T cells refines their prognostic utility for pancreatic cancer survival. Mod. Pathol. 2019, 32, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Erbe, R.; Danilova, L.; Phyo, Z.; Bigelow, E.; Stein-O’Brien, G.; Thomas, D.L., 2nd; Charmsaz, S.; Gross, N.; Woolman, S.; et al. Multi-omic profiling of lung and liver tumor microenvironments of metastatic pancreatic cancer reveals site-specific immune regulatory pathways. Genome Biol. 2021, 22, 154. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Feldmann, K.; Maurer, C.; Peschke, K.; Teller, S.; Schuck, K.; Steiger, K.; Engleitner, T.; Ollinger, R.; Nomura, A.; Wirges, N.; et al. Mesenchymal Plasticity Regulated by Prrx1 Drives Aggressive Pancreatic Cancer Biology. Gastroenterology 2021, 160, 346–361.e324. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, Y.; Gallegos, V.; Sorrelle, N.; Zaid, M.M.; Toombs, J.; Du, W.; Wright, S.; Hagopian, M.; Wang, Z.; et al. Targeting TGFbetaR2-mutant tumors exposes vulnerabilities to stromal TGFbeta blockade in pancreatic cancer. EMBO Mol. Med. 2019, 11, e10515. [Google Scholar] [CrossRef]
- Vaughan, M.B.; Howard, E.W.; Tomasek, J.J. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell Res. 2000, 257, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1beta Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejarano, L.; Jordao, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Pitarresi, J.R.; Cuitino, M.C.; Kladney, R.D.; Woelke, S.A.; Sizemore, G.M.; Nayak, S.G.; Egriboz, O.; Schweickert, P.G.; Yu, L.; et al. Genetic ablation of Smoothened in pancreatic fibroblasts increases acinar-ductal metaplasia. Genes Dev. 2016, 30, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer-clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Johnson, B.A., 3rd; Yarchoan, M.; Lee, V.; Laheru, D.A.; Jaffee, E.M. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin. Cancer Res. 2017, 23, 1656–1669. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, V.P.; Luksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Maggio, F.; El-Shakankery, K.H. Desmoplasia and Biophysics in Pancreatic Ductal Adenocarcinoma: Can We Learn from Breast Cancer? Pancreas 2020, 49, 313–325. [Google Scholar] [CrossRef]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef] [Green Version]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef] [PubMed]
- Masugi, Y.; Abe, T.; Yamazaki, K.; Ueno, A.; Sakamoto, M. Three Distinct Stroma Types in Human Pancreatic Cancer Identified by Image Analysis of Fibroblast Subpopulations and Collagen-Response. Clin. Cancer Res. 2022, 28, 427. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Moresco, P.; Yan, R.; Li, J.; Gao, Y.; Biasci, D.; Yao, M.; Pearson, J.; Hechtman, J.F.; Janowitz, T.; et al. Carcinomas assemble a filamentous CXCL12-keratin-19 coating that suppresses T cell-mediated immune attack. Proc. Natl. Acad. Sci. USA 2022, 119, e2119463119. [Google Scholar] [CrossRef]
- Grunwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592.e5518. [Google Scholar] [CrossRef]
- Watanabe, K.; Mitsunaga, S.; Kojima, M.; Suzuki, H.; Irisawa, A.; Takahashi, H.; Sasaki, M.; Hashimoto, Y.; Imaoka, H.; Ohno, I.; et al. The “histological replacement growth pattern” represents aggressive invasive behavior in liver metastasis from pancreatic cancer. Cancer Med. 2020, 9, 3130–3141. [Google Scholar] [CrossRef]
- McDonald, O.G. The biology of pancreatic cancer morphology. Pathology 2022, 54, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Lenk, L.; Pein, M.; Will, O.; Gomez, B.; Viol, F.; Hauser, C.; Egberts, J.H.; Gundlach, J.P.; Helm, O.; Tiwari, S.; et al. The hepatic microenvironment essentially determines tumor cell dormancy and metastatic outgrowth of pancreatic ductal adenocarcinoma. Oncoimmunology 2017, 7, e1368603. [Google Scholar] [CrossRef] [PubMed]
- Maddipati, R.; Stanger, B.Z. Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov. 2015, 5, 1086–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef] [PubMed]
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 2020, 10, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, T.; Mizutani, Y.; Esaki, N.; Ponik, S.M.; Burkel, B.M.; Weng, L.; Kuwata, K.; Masamune, A.; Ishihara, S.; Haga, H.; et al. Pharmacologic conversion of cancer-associated fibroblasts from a protumor phenotype to an antitumor phenotype improves the sensitivity of pancreatic cancer to chemotherapeutics. Oncogene 2022, 41, 2764–2777. [Google Scholar] [CrossRef]
- Franco-Barraza, J.; Francescone, R.; Luong, T.; Shah, N.; Madhani, R.; Cukierman, G.; Dulaimi, E.; Devarajan, K.; Egleston, B.L.; Nicolas, E.; et al. Matrix-regulated integrin alphavbeta5 maintains alpha5beta1-dependent desmoplastic traits prognostic of neoplastic recurrence. eLife 2017, 6, e20600. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Liu, Q.; Wu, J.; Xu, B.; Wang, J.; Liang, L.; Guo, Y.; Peng, M.; Zhao, Y.; Liao, Q. Fibroblast activation protein alpha-positive pancreatic stellate cells promote the migration and invasion of pancreatic cancer by CXCL1-mediated Akt phosphorylation. Ann. Transl. Med. 2019, 7, 532. [Google Scholar] [CrossRef]
- Hasselluhn, M.C.; Klein, L.; Patzak, M.S.; Buchholz, S.M.; Strobel, P.; Ellenrieder, V.; Maisonneuve, P.; Neesse, A. Stromal Features of the Primary Tumor Are Not Prognostic in Genetically Engineered Mice of Pancreatic Cancer. Cells 2019, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Lee, Y.; Lee, H.; Kim, J.W.; Hwang, J.H.; Kim, J.; Yoon, Y.S.; Han, H.S.; Kim, H. The prognostic significance of cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Tumour Biol. 2017, 39, 1010428317718403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawase, T.; Yasui, Y.; Nishina, S.; Hara, Y.; Yanatori, I.; Tomiyama, Y.; Nakashima, Y.; Yoshida, K.; Kishi, F.; Nakamura, M.; et al. Fibroblast activation protein-alpha-expressing fibroblasts promote the progression of pancreatic ductal adenocarcinoma. BMC Gastroenterol. 2015, 15, 109. [Google Scholar] [CrossRef] [Green Version]
- Bever, K.M.; Sugar, E.A.; Bigelow, E.; Sharma, R.; Laheru, D.; Wolfgang, C.L.; Jaffee, E.M.; Anders, R.A.; De Jesus-Acosta, A.; Zheng, L. The prognostic value of stroma in pancreatic cancer in patients receiving adjuvant therapy. HPB 2015, 17, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinn, M.; Denkert, C.; Striefler, J.K.; Pelzer, U.; Stieler, J.M.; Bahra, M.; Lohneis, P.; Dorken, B.; Oettle, H.; Riess, H.; et al. alpha-Smooth muscle actin expression and desmoplastic stromal reaction in pancreatic cancer: Results from the CONKO-001 study. Br. J. Cancer 2014, 111, 1917–1923. [Google Scholar] [CrossRef] [Green Version]
- Fujita, H.; Ohuchida, K.; Mizumoto, K.; Nakata, K.; Yu, J.; Kayashima, T.; Cui, L.; Manabe, T.; Ohtsuka, T.; Tanaka, M. α-Smooth Muscle Actin Expressing Stroma Promotes an Aggressive Tumor Biology in Pancreatic Ductal Adenocarcinoma. Pancreas 2010, 39, 1254–1262. [Google Scholar] [CrossRef]
- Cohen, S.J.; Alpaugh, R.K.; Palazzo, I.; Meropol, N.J.; Rogatko, A.; Xu, Z.; Hoffman, J.P.; Weiner, L.M.; Cheng, J.D. Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas 2008, 37, 154–158. [Google Scholar] [CrossRef]
- Mahajan, U.M.; Langhoff, E.; Goni, E.; Costello, E.; Greenhalf, W.; Halloran, C.; Ormanns, S.; Kruger, S.; Boeck, S.; Ribback, S.; et al. Immune Cell and Stromal Signature Associated with Progression-Free Survival of Patients With Resected Pancreatic Ductal Adenocarcinoma. Gastroenterology 2018, 155, 1625–1639.e1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2015, 28, 831–833. [Google Scholar] [CrossRef] [Green Version]
- McAndrews, K.M.; Chen, Y.; Darpolor, J.K.; Zheng, X.; Yang, S.; Carstens, J.L.; Li, B.; Wang, H.; Miyake, T.; Correa de Sampaio, P.; et al. Identification of Functional Heterogeneity of Carcinoma-Associated Fibroblasts with Distinct IL-6 Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discov. 2022, 12, 1580–1597. [Google Scholar] [CrossRef]
- Erkan, M.; Michalski, C.W.; Rieder, S.; Reiser-Erkan, C.; Abiatari, I.; Kolb, A.; Giese, N.A.; Esposito, I.; Friess, H.; Kleeff, J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2008, 6, 1155–1161. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565.e546. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Hamberger, F.; Ravichandra, A.; Miller, M.; Nair, A.; Affo, S.; Filliol, A.; Chin, L.; Savage, T.M.; Yin, D.; et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J. Clin. Investig. 2021, 131, e146987. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Target | Candidate Drug and Combination Regimen | Drug Type | Mechanism | Outcomes |
|---|---|---|---|---|
| LOXL2 | Simtuzumab plus gemcitabine | Blocking antibody | Destabilizes collagen networks | Negative outcome (phase II [58]) |
| Renin−angiotensin system | Losartan plus FOLFIRINOX followed by chemoradiotherapy (as neoadjuvant therapy) | Small molecule inhibitor | Reduces collagen and hyaluronan | Downstaging (phase II [59]) |
| Focal adhesion kinases | Defactinib plus pembrolizumab and gemcitabine | Small molecule inhibitor | Prevents integrin signaling | Clinical trials in phase I completed ([60], NCT02546531) |
| Hyaluronan | PEGPH20 plus nab-paclitaxel and gemcitabine | Enzyme | Degrades hyaluronan | Improved PFS (phase II [61]) |
| PEGPH20 plus modified FOLFIRINOX | Detrimental effects (phase Ib/II [62]) | |||
| PEGPH20 plus nab-paclitaxel and gemcitabine | Negative outcome (phase III [63]) | |||
| Hedgehog | Saridegib plus gemcitabine | Small molecule inhibitor | Prevents/reduces CAF activation | Worse clinical outcome (phase Ib/II, NCT01130142) |
| CXCR4-CXCL12 | Motixafortide plus pembrolizumab and chemotherapy | Small molecule inhibitor | Interferes with CAF signaling | Improved objective response rate (phase II [64,65]) |
| FAP | RO6874281 | Small molecule inhibitor | Interferes with CAF function | Clinical trials in phase I ongoing (NCT02627274) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masugi, Y. The Desmoplastic Stroma of Pancreatic Cancer: Multilayered Levels of Heterogeneity, Clinical Significance, and Therapeutic Opportunities. Cancers 2022, 14, 3293. https://doi.org/10.3390/cancers14133293
Masugi Y. The Desmoplastic Stroma of Pancreatic Cancer: Multilayered Levels of Heterogeneity, Clinical Significance, and Therapeutic Opportunities. Cancers. 2022; 14(13):3293. https://doi.org/10.3390/cancers14133293
Chicago/Turabian StyleMasugi, Yohei. 2022. "The Desmoplastic Stroma of Pancreatic Cancer: Multilayered Levels of Heterogeneity, Clinical Significance, and Therapeutic Opportunities" Cancers 14, no. 13: 3293. https://doi.org/10.3390/cancers14133293
APA StyleMasugi, Y. (2022). The Desmoplastic Stroma of Pancreatic Cancer: Multilayered Levels of Heterogeneity, Clinical Significance, and Therapeutic Opportunities. Cancers, 14(13), 3293. https://doi.org/10.3390/cancers14133293