
Stromal Co-Cultivation for Modeling Breast Cancer Dormancy in the Bone Marrow

{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Interactions of Disseminated Cancer Cells and Bone Marrow Cell Types in the Dormant Niche
3. Components of 2D In Vitro Dormancy Models
3.1. Mesenchymal Stem Cells
3.2. Bone Marrow Stromal Fibroblasts
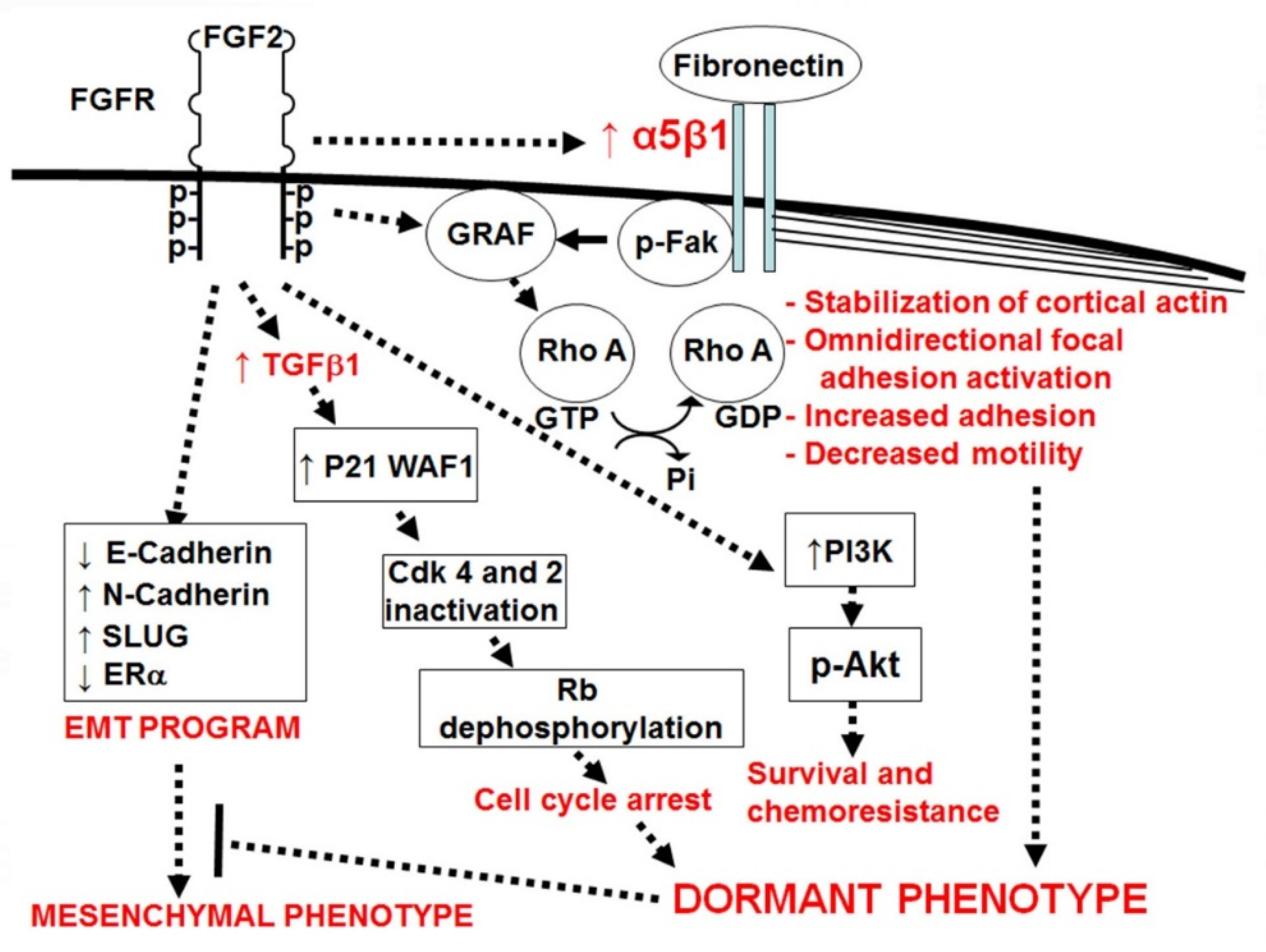
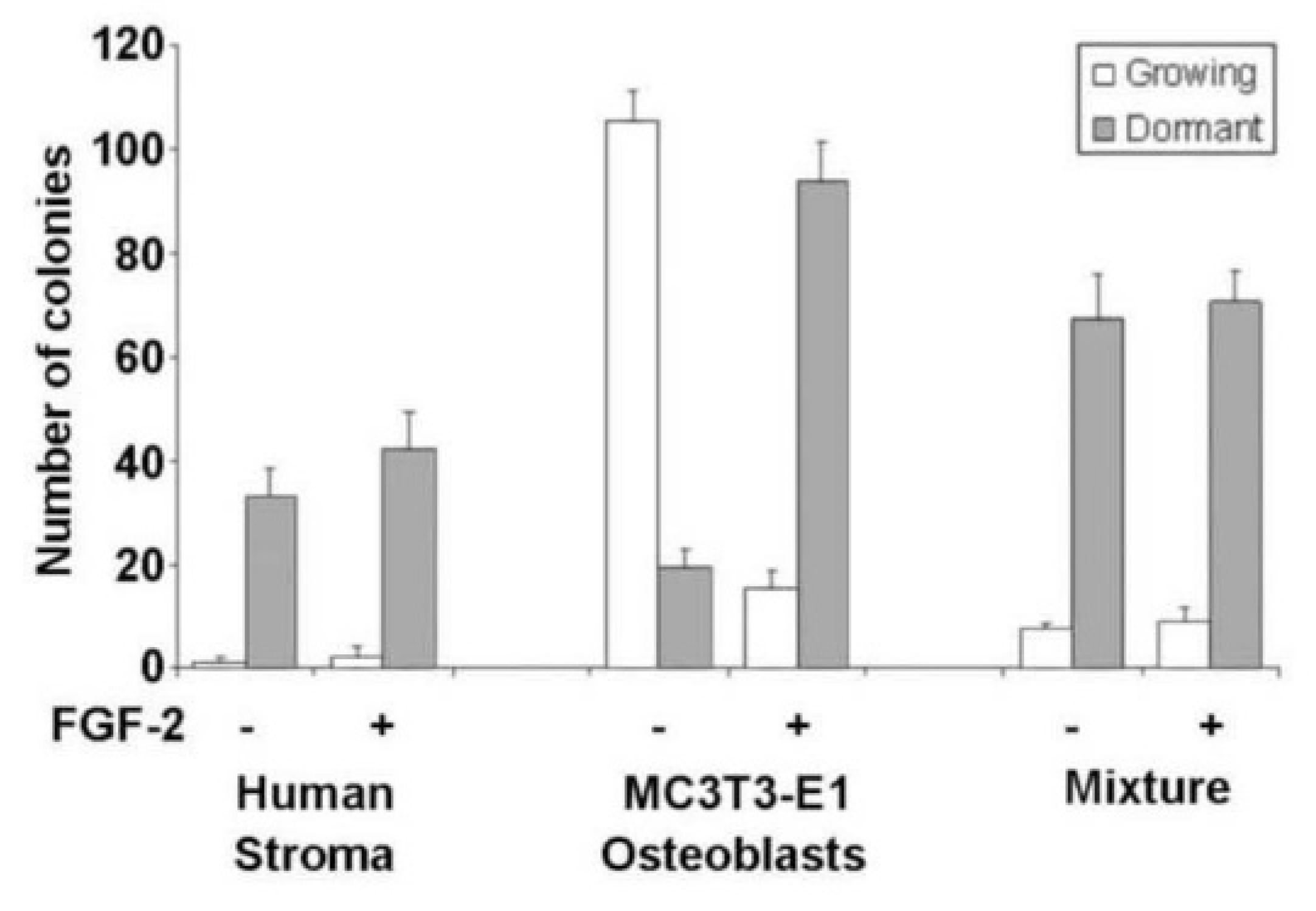
3.3. Osteoblasts
3.4. Adipocytes
3.5. Osteoclasts
3.6. Endothelial Cells
3.6.1. The Role of Endothelial Cells in Dormancy
3.6.2. Cancer Cells Injure Endothelial Cells
3.6.3. Endothelial Cells Modify Cancer Cells
3.6.4. Endothelial Cell Sources and Characteristics
3.6.5. Endothelial Cell Culture Conditions
3.6.6. Two-Dimensional Dormancy Cell–Cell Contact Co-Cultures
3.7. Other Components
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures. 2021. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2021.html (accessed on 19 October 2021).
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kittler, O.; Ragg, T.; Daskalakis, A.; Granzow, M.; Ahr, A.; Blankenstein, T.J.; Kaufmann, M.; Diebold, J.; Arnholdt, H.; Muller, P.; et al. From latent disseminated cells to overt metastasis: Genetic analysis of systemic breast cancer progression. Proc. Natl. Acad. Sci. USA 2003, 100, 7737–7742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, H.; Obradovic, M.M.S.; Hoffmann, M.; Harper, K.L.; Sosa, M.S.; Werner-Klein, M.; Nanduri, L.K.; Werno, C.; Ehrl, C.; Maneck, M.; et al. Early dissemination seeds metastasis in breast cancer. Nature 2016, 540, 552–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.A. Cancer progression and the invisible phase of metastatic colonization. Nat. Rev. Cancer 2020, 20, 681–694. [Google Scholar] [CrossRef]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef]
- Wieder, R. Insurgent micrometastases: Sleeper cells and harboring the enemy. J. Surg. Oncol. 2005, 89, 207–210. [Google Scholar] [CrossRef]
- Klein, C.A. Framework models of tumor dormancy from patient-derived observations. Curr. Opin. Genet. Dev. 2011, 21, 42–49. [Google Scholar] [CrossRef]
- Fang, C.; Kang, Y. Cellular Plasticity in Bone Metastasis. Bone 2020, 158, 115693. [Google Scholar] [CrossRef]
- Risson, E.; Nobre, A.R.; Maguer-Satta, V.; Aguirre-Ghiso, J.A. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat. Cancer 2020, 1, 672–680. [Google Scholar] [CrossRef]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for can-cer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Marlow, R.; Honeth, G.; Lombardi, S.; Cariati, M.; Hessey, S.; Pipili, A.; Mariotti, V.; Buchupalli, B.; Foster, K.; Bonnet, D.; et al. A novel model of dormancy for bone metastatic breast cancer cells. Cancer Res. 2013, 73, 6886–6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, M.; Saito, A.; Yamaguchi, Y.; Ohshima, M.; Nagase, T. Three-dimensional Co-culture model for tumor-stromal interac-tion. J. Vis. Exp. 2015, 2, 52469. [Google Scholar]
- Pradhan, S.; Sperduto, J.L.; Farino, C.J.; Slater, J.H. Engineered In Vitro Models of Tumor Dormancy and Reactivation. J. Biol. Eng. 2018, 12, 37. [Google Scholar] [CrossRef]
- Carpenter, R.A.; Kwak, J.G.; Peyton, S.R.; Lee, J. Implantable pre-metastatic niches for the study of the microenvironmental regulation of disseminated human tumour cells. Nat. Biomed. Eng. 2018, 2, 915–929. [Google Scholar] [CrossRef]
- Walker, C.; Mojares, E.; Del Rio Hernandez, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [Green Version]
- Carlson, P.; Dasgupta, A.; Grzelak, C.A.; Kim, J.; Barrett, A.; Coleman, I.M.; Shor, R.E.; Goddard, E.T.; Dai, J.; Schweitzer, E.M.; et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 2019, 21, 238–250. [Google Scholar] [CrossRef]
- McGrath, J.; Panzica, L.; Ransom, R.; Withers, H.G.; Gelman, I.H. Identification of Genes Regulating Breast Cancer Dormancy in 3D Bone Endosteal Niche Cultures. Mol. Cancer Res. MCR 2019, 17, 860–869. [Google Scholar] [CrossRef]
- Montagner, M.; Sahai, E. In vitro Models of Breast Cancer Metastatic Dormancy. Front. Cell Dev. Biol. 2020, 8, 37. [Google Scholar] [CrossRef] [Green Version]
- Mayhew, V.; Omokehinde, T.; Johnson, R.W. Tumor dormancy in bone. Cancer Rep. 2020, 3, e1156. [Google Scholar] [CrossRef]
- Dhurjati, R.; Liu, X.; Gay, C.V.; Mastro, A.M.; Vogler, E.A. Extended-term culture of bone cells in a compartmentalized bioreactor. Tissue Eng. 2006, 12, 3045–3054. [Google Scholar] [CrossRef] [PubMed]
- Sosnoski, D.M.; Norgard, R.J.; Grove, C.D.; Foster, S.J.; Mastro, A.M. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin. Exp. Metastasis 2015, 32, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, D.; Landau, S.; Shandalov, Y.; Raindel, N.; Freiman, A.; Shor, E.; Blinder, Y.; Vandenburgh, H.H.; Mooney, D.J.; Levenberg, S. Morphogenesis of 3D vascular networks is regulated by tensile forces. Proc. Natl. Acad. Sci. USA 2016, 113, 3215–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinisch, A.; Hernandez, D.C.; Schallmoser, K.; Majeti, R. Generation and use of a humanized bone-marrow-ossicle niche for hematopoietic xenotransplantation into mice. Nat. Protoc. 2017, 12, 2169–2188. [Google Scholar] [PubMed]
- Vanderburgh, J.P.; Guelcher, S.A.; Sterling, J.A. 3D bone models to study the complex physical and cellular interactions be-tween tumor and the bone microenvironment. J. Cell. Biochem. 2018, 119, 5053–5059. [Google Scholar] [CrossRef] [PubMed]
- Montagner, M.; Dupont, S. Mechanical Forces as Determinants of Disseminated Metastatic Cell Fate. Cells 2020, 9, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sistigu, A.; Musella, M.; Galassi, C.; Vitale, I.; De Maria, R. Tuning Cancer Fate: Tumor Microenvironment’s Role in Cancer Stem Cell Quiescence and Reawakening. Front. Immunol. 2020, 11, 2166. [Google Scholar] [CrossRef]
- Peister, A.; Mellad, J.A.; Larson, B.L.; Hall, B.M.; Gibson, L.F.; Prockop, D.J. Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood 2004, 103, 1662–1668. [Google Scholar] [CrossRef] [Green Version]
- Abou Nader, Z.; Espéli, M.; Balabanian, K.; Lemos, J.P. Culture, Expansion and Differentiation of Mouse Bone-Derived Mes-enchymal Stromal Cells. Methods Mol. Biol. 2021, 2308, 35–46. [Google Scholar]
- Bisio, V.; Espeli, M.; Balabanian, K.; Anginot, A. Culture, Expansion and Differentiation of Human Bone Marrow Stromal Cells. Methods Mol. Biol. 2021, 2308, 3–20. [Google Scholar]
- Ridge, S.M.; Sullivan, F.J.; Glynn, S.A. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, K.; Guezguez, B. Dynamic Changes of the Bone Marrow Niche: Mesenchymal Stromal Cells and Their Progeny During Aging and Leukemia. Front. Cell Dev. Biol. 2021, 9, 714716. [Google Scholar] [CrossRef] [PubMed]
- Czernik, M.; Fidanza, A.; Sardi, M.; Galli, C.; Brunetti, D.; Malatesta, D.; Della Salda, L.; Matsukawa, K.; Ptak, G.E.; Loi, P. Differentiation potential and GFP labeling of sheep bone marrow-derived mesenchymal stem cells. J. Cell. Biochem. 2013, 114, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.J.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Ther-apy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Abdallah, B.M.; Alzahrani, A.M.; Abdel-Moneim, A.M.; Ditzel, N.; Kassem, M. A simple and reliable protocol for long-term culture of murine bone marrow stromal (mesenchymal) stem cells that retained their in vitro and in vivo stemness in long-term culture. Biol. Proced. Online 2019, 21, 3. [Google Scholar] [CrossRef]
- Kafienah, W.; Mistry, S.; Williams, C.; Hollander, A.P. Nucleostemin is a marker of proliferating stromal stem cells in adult human bone marrow. Stem Cells 2006, 24, 1113–1120. [Google Scholar] [CrossRef]
- Li, H.; Liu, P.; Xu, S.; Li, Y.; Dekker, J.D.; Li, B.; Fan, Y.; Zhang, Z.; Hong, Y.; Yang, G.; et al. FOXP1 controls mesenchymal stem cell commitment and senescence during skeletal aging. J. Clin. Investig. 2017, 127, 1241–1253. [Google Scholar] [CrossRef]
- Wolf, N.S.; Penn, P.E.; Rao, D.; McKee, M.D. Intraclonal plasticity for bone, smooth muscle, and adipocyte lineages in bone marrow stroma fibroblastoid cells. Exp. Cell Res. 2003, 290, 346–357. [Google Scholar] [CrossRef]
- Bae, S.H.; Ryu, H.; Rhee, K.J.; Oh, J.E.; Baik, S.K.; Shim, K.Y.; Kong, J.H.; Hyun, S.Y.; Pack, H.S.; Im, C.; et al. L-ascorbic acid 2-phosphate and fibroblast growth factor-2 treatment maintains differentiation potential in bone marrow-derived mesen-chymal stem cells through expression of hepatocyte growth factor. Growth Factors 2015, 33, 71–78. [Google Scholar] [CrossRef]
- Baddoo, M.; Hill, K.; Wilkinson, R.; Gaupp, D.; Hughes, C.; Kopen, G.C.; Phinney, D.G. Characterization of mesenchymal stem cells isolated from murine bone marrow by negative selection. J. Cell. Biochem. 2003, 89, 1235–1249. [Google Scholar] [CrossRef]
- Sun, S.; Guo, Z.; Xiao, X.; Liu, B.; Liu, X.; Tang, P.H.; Mao, N. Isolation of mouse marrow mesenchymal progenitors by a novel and reliable method. Stem Cells 2003, 21, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.; Boucher, S.; Koh, S.; Sastry, K.S.R.; Chase, L.; Lakshmipathy, U.; Choong, C.; Yang, Z.; Vemuri, M.C.; Rao, M.S.; et al. PDGF, TGF-β, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): Tran-scriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood 2008, 112, 295–307. [Google Scholar]
- Coutu, D.L.; Galipeau, J. Roles of FGF signaling in stem cell self-renewal, senescence and aging. Aging 2011, 3, 920–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caroti, C.M.; Ahn, H.; Salazar, H.F.; Joseph, G.; Sankar, S.B.; Willett, N.J.; Wood, L.B.; Taylor, W.R.; Lyle, A.N. A novel technique for accelerated culture of murine mesenchymal stem cells that allows for sustained multipotency. Sci. Rep. 2017, 7, 13334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Son, W.S.; Park, M.C.; Kim, C.M.; Cha, B.H.; Yoon, K.J.; Lee, S.H.; Park, S.G. ARS-interacting multi-functional protein 1 induces proliferation of human bone marrow-derived mesenchymal stem cells by accumulation of beta-catenin via fibroblast growth factor receptor 2-mediated activation of Akt. Stem Cells Dev. 2013, 22, 2630–2640. [Google Scholar] [CrossRef] [Green Version]
- Huh, J.E.; Ko, R.; Jung, H.J.; Lee, S.Y. Glycogen synthase kinase 3beta promotes osteogenic differentiation of murine adi-pose-derived stromal cells. PLoS ONE 2013, 8, e54551. [Google Scholar]
- Ishikane, S.; Ikushima, E.; Igawa, K.; Tomooka, K.; Takahashi-Yanaga, F. Differentiation-inducing factor-1 potentiates adi-pogenic differentiation and attenuates the osteogenic differentiation of bone marrow-derived mesenchymal stem cells. Bio-chim. Biophys. Acta-Mol. Cell Res. 2021, 1868, 118909. [Google Scholar]
- Baksh, D.; Tuan, R.S. Canonical and non-canonical Wnts differentially affect the development potential of primary isolate of human bone marrow mesenchymal stem cells. J. Cell. Physiol. 2007, 212, 817–826. [Google Scholar] [CrossRef]
- Ito, T.; Sawada, R.; Fujiwara, Y.; Tsuchiya, T. FGF-2 increases osteogenic and chondrogenic differentiation potentials of human mesenchymal stem cells by inactivation of TGF-beta signaling. Cytotechnology 2008, 56, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Handorf, A.M.; Li, W.J. Fibroblast growth factor-2 primes human mesenchymal stem cells for enhanced chondrogenesis. PLoS ONE 2011, 6, e22887. [Google Scholar] [CrossRef] [Green Version]
- Kwan, M.D.; Sellmyer, M.A.; Quarto, N.; Ho, A.M.; Wandless, T.J.; Longaker, M.T. Chemical control of FGF-2 release for promoting calvarial healing with adipose stem cells. J. Biol. Chem. 2011, 286, 11307–11313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Niu, J.; Li, X.; Wang, X.; Guo, Z.; Zhang, F. TGF-beta1 induces senescence of bone marrow mesenchymal stem cells via increase of mitochondrial ROS production. BMC Dev. Biol. 2014, 14, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakudo, N.; Shimotsuma, A.; Kusumoto, K. Fibroblast growth factor-2 stimulates adipogenic differentiation of human adi-pose-derived stem cells. Biochem. Biophys. Res. Commun. 2007, 359, 239–244. [Google Scholar] [CrossRef]
- Hutley, L.J.; Newell, F.S.; Kim, Y.H.; Luo, X.; Widberg, C.H.; Shurety, W.; Prins, J.B.; Whitehead, J.P. A putative role for en-dogenous FGF-2 in FGF-1 mediated differentiation of human preadipocytes. Mol. Cell. Endocrinol. 2011, 339, 165–171. [Google Scholar] [CrossRef]
- Patel, N.G.; Kumar, S.; Eggo, M.C. Essential role of fibroblast growth factor signaling in preadipoctye differentiation. J. Clin. Endocrinol. Metab. 2005, 90, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Prusty, D.; Park, B.H.; Davis, K.E.; Farmer, S.R. Activation of MEK/ERK signaling promotes adipogenesis by enhancing pe-roxisome proliferator-activated receptor gamma (PPARgamma ) and C/EBPalpha gene expression during the differentiation of 3T3-L1 preadipocytes. J. Biol. Chem. 2002, 277, 46226–46232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Shah, B.; Moioli, E.K.; Mao, J.J. CTGF directs fibroblast differentiation from human mesenchymal stem/stromal cells and defines connective tissue healing in a rodent injury model. J. Clin. Investig. 2010, 120, 3340–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadie-Van Gijsen, H.; Crowther, N.J.; Hough, F.S.; Ferris, W.F. The interrelationship between bone and fat: From cellular see-saw to endocrine reciprocity. Cell. Mol. Life Sci. 2013, 70, 2331–2349. [Google Scholar] [CrossRef]
- Balani, D.H.; Ono, N.; Kronenberg, H.M. Parathyroid hormone regulates fates of murine osteoblast precursors in vivo. J. Clin. Investig. 2017, 127, 3327–3338. [Google Scholar] [CrossRef] [Green Version]
- Seok, J.W.; Kim, D.; Yoon, B.K.; Lee, Y.; Kim, H.J.; Hwang, N.; Fang, S.; Kim, H.J.; Kim, J.W. Dexras1 plays a pivotal role in maintaining the equilibrium between adipogenesis and osteogenesis. Metab. Clin. Exp. 2020, 108, 154250. [Google Scholar] [CrossRef]
- Hashimoto, R.; Katoh, Y.; Miyamoto, Y.; Itoh, S.; Daida, H.; Nakazato, Y.; Okada, T. Increased extracellular and intracellular Ca2+ lead to adipocyte accumulation in bone marrow stromal cells by different mechanisms. Biochem. Biophys. Res. Commun. 2015, 457, 647–652. [Google Scholar] [CrossRef] [PubMed]
- De Winter, T.J.J.; Nusse, R. Running Against the Wnt: How Wnt/beta-Catenin Suppresses Adipogenesis. Front. Cell Dev. Biol. 2021, 9, 627429. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Jiang, S.; Hantash, B.M. Transforming growth factor beta1 induces osteogenic differentiation of murine bone mar-row stromal cells. Tissue Eng. Part A 2010, 16, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ruan, M.; Clifton, K.; Syed, F.; Khosla, S.; Oursler, M.J. TGF-beta mediates suppression of adipogenesis by estra-diol through connective tissue growth factor induction. Endocrinology 2012, 153, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Hesslein, D.G.; Fretz, J.A.; Xi, Y.; Nelson, T.; Zhou, S.; Lorenzo, J.A.; Schatz, D.G.; Horowitz, M.C. Ebf1-dependent control of the osteoblast and adipocyte lineages. Bone 2009, 44, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Prado-Lopez, S.; Duffy, M.M.; Baustian, C.; Alagesan, S.; Hanley, S.A.; Stocca, A.; Griffin, M.D.; Ceredig, R. The influence of hypoxia on the differentiation capacities and immunosuppressive properties of clonal mouse mesenchymal stromal cell lines. Immunol. Cell Biol. 2014, 92, 612–623. [Google Scholar] [CrossRef]
- Kakudo, N.; Morimoto, N.; Ogawa, T.; Taketani, S.; Kusumoto, K. Hypoxia Enhances Proliferation of Human Adi-pose-Derived Stem Cells via HIF-1a Activation. PLoS ONE 2015, 10, e0139890. [Google Scholar] [CrossRef] [Green Version]
- Griner, J.D.; Rogers, C.J.; Zhu, M.J.; Du, M. Lysyl oxidase propeptide promotes adipogenesis through inhibition of FGF-2 sig-naling. Adipocyte 2017, 6, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Velickovic, K.; Lugo Leija, H.A.; Bloor, I.; Law, J.; Sacks, H.; Symonds, M.; Sottile, V. Low temperature exposure induces browning of bone marrow stem cell derived adipocytes in vitro. Sci. Rep. 2018, 8, 4974. [Google Scholar] [CrossRef]
- Hu, L.; Yin, C.; Zhao, F.; Ali, A.; Ma, J.; Qian, A. Mesenchymal Stem Cells: Cell Fate Decision to Osteoblast or Adipocyte and Application in Osteoporosis Treatment. Int. J. Mol. Sci. 2018, 19, 360. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, M.; Nishikawa, S.; Ikuta, K.; Yamamura, F.; Naito, M.; Takahashi, K.; Nishikawa, S. B cell ontogeny in murine embryo studied by a culture system with the monolayer of a stromal cell clone, ST2: B cell progenitor develops first in the embryonal body rather than in the yolk sac. EMBO J. 1988, 7, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Yamaguchi, A.; Ikeda, T.; Yoshiki, S.; Wozney, J.M.; Rosen, V.; Wang, E.A.; Tanaka, H.; Omura, S.; Suda, T. The non-osteogenic mouse pluripotent cell line, C3H10T1/2, is induced to differentiate into osteoblastic cells by recombinant human bone morphogenetic protein. Biochem. Biophys. Res. Commun. 1990, 172, 295–299. [Google Scholar] [CrossRef]
- Negishi, Y.; Kudo, A.; Obinata, A.; Kawashima, K.; Hirano, H.; Yanai, N.; Obinata, M.; Endo, H. Multipotency of a bone marrow stromal cell line, TBR31-2, established from ts-SV40 T antigen gene transgenic mice. Biochem. Biophys. Res. Commun. 2000, 268, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, K.; Mayer, B.; Sworder, B.J.; Kuznetsov, S.A.; Mezey, E. A practical guide to culturing mouse and human bone marrow stromal cells. Curr. Protoc. Immunol. 2013, 102, 12.1–12.3. [Google Scholar] [CrossRef] [PubMed]
- Tivari, S.; Lu, H.; Dasgpta, T.; De Lorenzo, M.S.; Wieder, R. Reawakening of dormant estrogen-dependent human breast cancer cells by bone marrow stroma secretory senescence. Cell Commun. Signal. 2018, 16, 48. [Google Scholar] [CrossRef] [Green Version]
- Oliver, L.J.; Rifkin, D.B.; Gabrilove, J.L.; Hannocks, M.-J.; Wilson, E.L. Long-term culture of human bone marrow stromal cells in the presence of basic fibroblast growth factor. Growth Factors 1990, 3, 231–236. [Google Scholar] [CrossRef]
- Dobson, K.R.; Reading, L.; Haberey, M.; Marine, X.; Scutt, A. Centrifugal isolation of bone marrow from bone: An improved method for the recovery and quantification of bone marrow osteoprogenitor cells from rat tibiae and femurae. Calcif. Tissue Int. 1999, 65, 411–413. [Google Scholar] [CrossRef]
- Korah, R.; Boots, M.; Wieder, R. Integrin α5β1 promotes survival of growth-arrested breast cancer cells: An in vitro paradigm for breast cancer dormancy in bone marrow. Cancer Res. 2004, 64, 4514–4522. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.Y.; Chi, J.T.; Dudoit, S.; Bondre, C.; van de Rijn, M.; Botstein, D.; Brown, P.O. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 12877–12882. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.L.; Gardner, T.A.; Miao, L.; Balian, G.; Chung, L.W. Cotargeting tumor and stroma in a novel chimeric tumor model involving the growth of both human prostate cancer and bone stromal cells. Cancer Gene Ther. 2004, 11, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Kosala, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, S.J.; Rameshwar, P. Analysis of the transfer of circulating microRNA between cells mediated by gap junction. Methods Mol. Biol. 2013, 1024, 87–96. [Google Scholar]
- Ovadia, E.M.; Pradhan, L.; Sawicki, L.A.; Cowart, J.E.; Huber, R.E.; Polson, S.W.; Chen, C.; van Golen, K.L.; Ross, K.E.; Wu, C.H.; et al. Understanding ER+ Breast Cancer Dormancy Using Bioinspired Synthetic Matrices for Long-Term 3D Culture and Insights into Late Recurrence. Adv. Biosyst. 2020, 4, e2000119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zuo, D.; Wang, M.; Zhang, Y.; Yu, M.; Yang, J.; Yao, Z. Effect of truncated neurokinin-1 receptor expression changes on the interaction between human breast cancer and bone marrow-derived mesenchymal stem cells. Genes Cells 2014, 19, 676–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, G.; Nguyen, H.; Gabrilove, J.; Rifkin, D.B.; Wilson, E.L. Basic fibroblast growth factor expression in human bone marrow and peripheral blood cells. Blood 1993, 81, 631–638. [Google Scholar]
- Nilsson, S.K.; Debatis, M.E.; Dooner, M.S.; Madri, J.A.; Quesenberry, P.J.; Becker, P.S. Immunofluorescence characterization of key extracellular matrix proteins in murine bone marrow in situ. J. Histochem. Cytochem. 1998, 46, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Barney, L.E.; Hall, C.L.; Schwartz, A.D.; Parks, A.N.; Sparages, C.; Galarza, S.; Platt, M.O.; Mercurio, A.M.; Peyton, S.R. Tumor cell-organized fibronectin maintenance of a dormant breast cancer population. Sci. Adv. 2020, 6, eaaz4157. [Google Scholar] [CrossRef] [Green Version]
- Giancotti, F.G.; Ruoslahti, E. Elevated levels of the alpha 5 beta 1 fibronectin receptor suppress the transformed phenotype of Chinese hamster ovary cells. Cell 1990, 60, 849–859. [Google Scholar] [CrossRef]
- Giancotti, F.G. Mechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef] [Green Version]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrios, J.; Wieder, R. Dual FGF-2 and intergrin α5β1 signaling mediate GRAF-induced RhoA inactivation in a model of breast cancer dormancy. Cancer Microenviron. 2009, 2, 33–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tivari, S.; Korah, R.; Lindy, M.; Wieder, R. An in vitro dormancy model of estrogen-sensitive breast cancer in the bone marrow: A tool for molecular mechanism studies and hypothesis generation. J. Vis. Exp. 2015, 100, e52672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najmi, S.; Korah, R.; Chandra, R.; Abdellatif, M.; Wieder, R. Flavopiridol blocks integrin-mediated survival in dormant breast cancer cells. Clin. Cancer Res. 2005, 11, 2038–2046. [Google Scholar] [CrossRef] [Green Version]
- Fenig, E.; Wieder, R.; Paglin, S.; Wang, H.; Persaud, R.; Haimovitz-Friedman, A.; Fuks, Z.; Yahalom, J. Basic fibroblast growth factor confers growth inhibition and Mitogen-activated Protein Kinase activation in human breast cancer cells. Clin. Cancer Res. 1997, 3, 135–142. [Google Scholar]
- Wang, H.; Rubin, M.; Fenig, E.; DeBlasio, T.; Mendelsohn, J.; Yahalom, J.; Wieder, R. Basic FGF causes growth arrest in MCF-7 human breast cancer cells while inducing both mitogenic and inhibitory G1 events. Cancer Res. 1997, 57, 1750–1757. [Google Scholar]
- Fenig, E.; Kanfi, Y.; Wang, Q.; Beery, E.; Livnat, T.; Wasserman, L.; Lilling, G.; Yahalom, J.; Wieder, R.; Nordenberg, J. Role of transforming growth factor beta in the growth inhibition of human breast cancer cells by basic fibroblast growth factor. Breast Cancer Res. Treat. 2001, 70, 27–37. [Google Scholar] [CrossRef]
- Huang, J.; Woods, P.; Normolle, D.; Goff, J.P.; Benos, P.V.; Stehle, C.J.; Steinman, R.A. Downregulation of estrogen receptor and modulation of growth of breast cancer cell lines mediated by paracrine stromal cell signals. Breast Cancer Res. Treat. 2017, 161, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef]
- Pommier, A.; Anaparthy, N.; Memos, N.; Kelley, Z.L.; Gouronnec, A.; Yan, R.; Auffray, C.; Albrengues, J.; Egeblad, M.; Iacobuzio-Donahue, C.A.; et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 2018, 360, eaao4908. [Google Scholar] [CrossRef] [Green Version]
- Probert, C.; Dottorini, T.; Speakman, A.; Hunt, S.; Nafee, T.; Fazeli, A.; Wood, S.; Brown, J.E.; James, V. Communication of prostate cancer cells with bone cells via extracellular vesicle RNA; a potential mechanism of metastasis. Oncogene 2019, 38, 1751–1763. [Google Scholar] [CrossRef] [PubMed]
- Chiovaro, F.; Martina, E.; Bottos, A.; Scherberich, A.; Hynes, N.E.; Chiquet-Ehrismann, R. Transcriptional regulation of tenascin-W by TGF-beta signaling in the bone metastatic niche of breast cancer cells. Int. J. Cancer 2015, 137, 1842–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Tian, L.; Liu, J.; Goldstein, A.; Bado, I.; Zhang, W.; Arenkiel, B.R.; Li, Z.; Yang, M.; Du, S.; et al. The ostogenic niche is a calcium reservoir of bone micrometastases and conferes unexpected therapeutic vulnerability. Cancer Cell 2018, 34, 823–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Kamiya, S.; Akatsu, T.; Nakamura, C.; Li, M.; Amizuka, N.; Matsumoto, K.; Nakamura, T.; Kugai, N.; Wada, S. Involvement of hepatocyte growth factor in the development of bone metastasis of a mouse mammary cancer cell line, BALB/c-MC. Bone 2006, 39, 27–34. [Google Scholar] [CrossRef]
- Hughes, R.; Chen, X.; Cowley, N.; Ottewell, P.D.; Hawkins, R.J.; Hunter, K.D.; Hobbs, J.K.; Brown, N.J.; Holen, I. Osteo-blast-derived paracrine and juxtacrine signals protect disseminated breast cancer cells from stress. Cancers 2021, 13, 1366. [Google Scholar] [CrossRef]
- Luo, X.; Fu, Y.; Loza, A.J.; Murali, B.; Leahy, K.M.; Ruhland, M.K.; Gang, M.; Su, X.; Zamani, A.; Shi, Y.; et al. Stromal-Initiated Changes in the Bone Promote Metastatic Niche Development. Cell Rep. 2016, 14, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Morrison, C.; Mancini, S.; Cipollone, J.; Kappelhoff, R.; Roskelley, C.; Overall, C. Microarray and proteomic analysis of breast cancer cell and osteoblast co-cultures: Role of osteoblast matrix metalloproteinase (MMP)-13 in bone metastasis. J. Biol. Chem. 2011, 286, 34271–34285. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, M.; Murata, T.; Shoji, M.; Weitzmann, M.N. The flavonoid p-hydroxycinnamic acid mediates anticancer effects on MDA-MB-231 human breast cancer cells in vitro: Implications for suppression of bone metastases. Int. J. Oncol. 2015, 47, 1563–1571. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, Y.; Havens, A.M.; Zalucha, S.; Ziegler, A.M.; Pederson, E.M.; Wang, Y.; Jung, Y.; Wang, J.H.; Ying, C.; Wang, J.; et al. Prostate Cancer Cells as Parasites of the Hematopoietic Stem Cell Niche. In Proceedings of the Skeletal Complications of Malignancy, Symposium of the Paget Foundation and the University of Michigan, Philadelphia, PA, USA, 25–27 October 2007; pp. 20, S21. [Google Scholar]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cackowski, F.C.; Taichman, R.S. Parallels between hematopoietic stem cell and prostate cancer disseminated tumor cell regulation. Bone 2019, 119, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Dominguez, A.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef]
- Capulli, M.; Hristova, D.; Valbret, Z.; Carys, K.; Arjan, R.; Maurizi, A.; Masedu, F.; Cappariello, A.; Rucci, N.; Teti, A. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br. J. Cancer 2019, 121, 157–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhang, W.; Bado, I.; Zhang, X.H.-F. Bone tropism in cancer metastases. Cold Spring Harb. Perspect. Med. 2020, 10, a036848. [Google Scholar] [CrossRef]
- Orriss, I.R.; Knight, G.E.; Ranasinghe, S.; Burnstock, G.; Arnett, T.R. Osteoblast responses to nucleotides increase during differentiation. Bone 2006, 39, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, B.M. Marrow adipocytes inhibit the differentiation of mesenchymal stem cells into osteoblasts via suppressing BMP-signaling. J. Biomed. Sci. 2017, 24, 11. [Google Scholar] [CrossRef] [Green Version]
- Doolittle, M.L.; Ackert-Bicknell, C.L.; Jonason, J.H. Isolation and Culture of Neonatal Mouse Calvarial Osteoblasts. Methods Mol. Biol. 2021, 2230, 425–436. [Google Scholar]
- Wang, W.; Majihail, G.; Lui, C.; Zhou, L. Osteoblast Sorting and Intracellular Staining of CXCL. Bio-Protoc. 2018, 8, 10. [Google Scholar] [CrossRef]
- Petecchia, L.; Viti, F.; Sbrana, F.; Vassalli, M.; Gavazzo, P. A biophysical approach to quantify skeletal stem cells trans-differentiation as a model for the study of osteoporosis. Biophys. Chem. 2017, 229, 84–92. [Google Scholar] [CrossRef]
- Xu, J.C.; Wu, G.H.; Zhou, L.L.; Yang, X.J.; Liu, J.T. Leptin improves osteoblast differentiation of human bone marrow stroma stem cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3507–3513. [Google Scholar] [PubMed]
- Gori, F.; Thomas, T.; Hicok, K.C.; Spelsberg, T.C.; Riggs, B.L. Differentiation of human marrow stromal precursor cells: Bone morphogenetic protein-2 increases OSF2/CBFA1, enhances osteoblast commitment, and inhibits late adipocyte maturation. J. Bone Miner. Res. 1999, 14, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Kolf, C.M.; Song, L.; Helm, J.; Tuan, R.S. Nascent osteoblast matrix inhibits osteogenesis of human mesenchymal stem cells in vitro. Stem Cell Res. Ther. 2015, 6, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, G.R., Jr.; Sullivan, E.C.; Moran, E.; Zerler, B. Relationship between alkaline phosphatase levels, osteopontin expression, and mineralization in differentiating MC3T3-E1 osteoblasts. J. Cell. Biochem. 1998, 68, 269–280. [Google Scholar] [CrossRef]
- Gregory, C.A.; Gunn, W.G.; Peister, A.; Prockop, D.J. An Alizarin red-based assay of mineralization by adherent cells in cul-ture: Comparison with cetylpyridinium chloride extraction. Anal. Biochem. 2004, 329, 77–84. [Google Scholar] [CrossRef]
- Atluri, K.; Seabold, D.; Hong, L.; Elangovan, S.; Salem, A.K. Nanoplex-Mediated Codelivery of Fibroblast Growth Factor and Bone Morphogenetic Protein Genes Promotes Osteogenesis in Human Adipocyte-Derived Mesenchymal Stem Cells. Mol. Pharm. 2015, 12, 3032–3042. [Google Scholar] [CrossRef] [Green Version]
- Sung, S.Y.; Hsieh, C.L.; Law, A.; Zhau, H.E.; Pathak, S.; Multani, A.S.; Lim, S.; Coleman, I.M.; Wu, L.C.; Figg, W.D.; et al. Coevolution of prostate cancer and bone stroma in three-dimensional coculture: Implications for cancer growth and metastasis. Cancer Res. 2008, 68, 9996–10003. [Google Scholar] [CrossRef] [Green Version]
- Bado, I.L.; Zhang, W.; Hu, J.; Xu, Z.; Wang, H.; Sarkar, P.; Li, L.; Wan, Y.W.; Liu, J.; Wu, W.; et al. The bone microenvironment increases phenotypic plasticity of ER+ breast cancer cells. Dev. Cell 2021, 56, 1100–1117.e9. [Google Scholar] [CrossRef]
- Kim, H.M.; Lee, Y.K.; Kim, E.S.; Koo, J.S. Energy transfer from adipocytes to cancer cells in breast cancer. Neoplasma 2020, 67, 992–1001. [Google Scholar] [CrossRef]
- Sadowski, H.B.; Wheeler, T.T.; Young, D.A. Gene expression during 3T3-L1 adipocyte differentiation. Characterization of initial responses to the inducing agents and changes during commitment to differentiation. J. Biol. Chem. 1992, 267, 4722–4731. [Google Scholar] [CrossRef]
- Meulle, A.; Salles, B.; Daviaud, D.; Valet, P.; Muller, C. Positive regulation of DNA double strand break repair activity during differentiation of long life span cells: The example of adipogenesis. PLoS ONE 2008, 3, e3345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Gan, L.; Ma, B.; He, S.; Wu, P.; Li, H.; Xiong, J. Alterations in chromatin accessibility during osteoblast and adipocyte differentiation in human mesenchymal stem cells. BMC Med. Genom. 2022, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Gesta, S.; Lolmede, K.; Daviaud, D.; Berlan, M.; Bouloumie, A.; Lafontan, M.; Valet, P.; Saulnier-Blache, J.S. Culture of hu-man adipose tissue explants leads to profound alteration of adipocyte gene expression. Horm. Metab. Res. 2003, 35, 158–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daviaud, D.; Boucher, J.; Gesta, S.; Dray, C.; Guigne, C.; Quilliot, D.; Ayav, A.; Ziegler, O.; Carpene, C.; Saulnier-Blache, J.S.; et al. TNFalpha up-regulates apelin expression in human and mouse adipose tissue. FASEB J. 2006, 20, 1528–1530. [Google Scholar] [CrossRef]
- Zhang, Y.; Daquinag, A.C.; Amaya-Manzanares, F.; Sirin, O.; Tseng, C.; Kolonin, M.G. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res. 2012, 72, 5198–5208. [Google Scholar] [CrossRef] [Green Version]
- Klopp, A.H.; Zhang, Y.; Solley, T.; Amaya-Manzanares, F.; Marini, F.; Andreeff, M.; Debeb, B.; Woodward, W.; Schmandt, R.; Broaddus, R.; et al. Omental adipose tissue-derived stromal cells promote vascularization and growth of endometrial tumors. Clin. Cancer Res. 2012, 18, 771–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Jung, W.H.; Koo, J.S. Adipocytes can induce epithelial-mesenchymal transition in breast cancer cells. Breast Can-cer Res. Treat. 2015, 153, 323–335. [Google Scholar] [CrossRef]
- Bochet, L.; Lehuede, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Go-nidec, S.; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Wang, D.; Feng, J.; Guo, P.; Geng, C. Denosumab inhibits MCF-7 cell line-induced spontaneous osteoclastogenesis via the RANKL/MALAT1/miR-124 axis. Transl. Cancer Res. 2020, 9, 2482–2491. [Google Scholar] [CrossRef]
- Mercatali, L.; La Manna, F.; Miserocchi, G.; Liverani, C.; De Vita, A.; Spadazzi, C.; Bongiovanni, A.; Recine, F.; Amadori, D.; Ghetti, M.; et al. Tumor-Stroma Crosstalk in Bone Tissue: The Osteoclastogenic Potential of a Breast Cancer Cell Line in a Co-Culture System and the Role of EGFR Inhibition. Int. J. Mol. Sci. 2017, 18, 1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benad-Mehner, P.; Thiele, S.; Rachner, T.D.; Gobel, A.; Rauner, M.; Hofbauer, L.C. Targeting syndecan-1 in breast cancer inhibits osteoclast functions through up-regulation of osteoprotegerin. J. Bone Oncol. 2014, 3, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, J.; Du, Q.; Hou, Y.; Wang, Z.; Zhang, J. Utilization of E-cadherin by monocytes from tumour cells plays key roles in the progression of bone invasion by oral squamous cell carcinoma. Oncol. Rep. 2017, 38, 850–858. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Yin, Y.; Yan, Z.; Cao, K.; Zhong, K. NAC1 promotes the migration of prostate cancer cells and participates in osteo-clastogenesis by negatively regulating IFNbeta. Oncol. Lett. 2018, 15, 2921–2928. [Google Scholar]
- Yang, Z.; Yue, Z.; Ma, X.; Xu, Z. Calcium Homeostasis: A Potential Vicious Cycle of Bone Metastasis in Breast Cancers. Front. Oncol. 2020, 10, 293. [Google Scholar] [CrossRef]
- Fujikawa, Y.; Quinn, J.M.; Sabokbar, A.; McGee, J.O.; Athanasou, N.A. The human osteoclast precursor circulates in the monocyte fraction. Endocrinology 1996, 137, 4058–4060. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.C.; Athanasou, N.A. Recent advances in osteoclast biology. Histol. Histopathol. 2004, 19, 189–199. [Google Scholar] [PubMed]
- Lau, Y.S.; Danks, L.; Sun, S.G.; Fox, S.; Sabokbar, A.; Harris, A.; Athanasou, N.A. RANKL-dependent and RANKL-independent mechanisms of macrophage-osteoclast differentiation in breast cancer. Breast Cancer Res. Treat. 2007, 105, 7–16. [Google Scholar] [CrossRef]
- Wada, A.; Tsuchiya, M.; Ozaki-Honda, Y.; Kayamori, K.; Sakamoto, K.; Yamaguchi, A.; Ikeda, T. A new osteoclastogenesis pathway induced by cancer cells targeting osteoclast precursor cells. Biochem. Biophys. Res. Commun. 2019, 509, 108–113. [Google Scholar] [CrossRef]
- Bugelski, P.J.; Kirsh, R.; Buscarino, C.; Corwin, S.P.; Poste, G. Recruitment of exogenous macrophages into metastases at dif-ferent stages of tumor growth. Cancer Immunol. Immunother. 1987, 24, 93–98. [Google Scholar] [CrossRef]
- Van Ravenswaay Classen, H.H.; Kluin, P.M.; Fleuren, G. Tumour infiltrating cells in human cancer: On the possible role of CD16+ macrophages in anti-tumour cytoxicity. Lab. Investig. 1992, 67, 166–174. [Google Scholar]
- Jin, W.J.; Kim, B.; Kim, D.; Park Choo, H.Y.; Kim, H.H.; Ha, H.; Lee, Z.H. NF-kappaB signaling regulates cell-autonomous regulation of CXCL10 in breast cancer 4T1 cells. Exp. Mol. Med. 2017, 49, e295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, T.; Kasai, M.; Suzuki, J.; Kuroyama, H.; Seki, S.; Utsuyama, M.; Hirokawa, K. Multimerization of the receptor activa-tor of nuclear factor-kappaB ligand (RANKL) isoforms and regulation of osteoclastogenesis. J. Biol. Chem. 2003, 278, 47217–47222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordstrand, A.; Nilsson, J.; Tieva, Å.; Wikström, P.; Lerner, U.H.; Widmark, A. Establishment and validation of an in vitro co-culture model to study the interactions between bone and prostate cancer cells. Clin. Exp. Metastasis 2009, 26, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Halade, G.V.; El Jamali, A.; Williams, P.J.; Fajardo, R.J.; Fernandes, G. Obesity-mediated inflammatory microenvironment stimulates osteoclastogenesis and bone loss in mice. Exp. Gerontol. 2011, 46, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Ho, L.; Wang, L.; Roth, T.M.; Pan, Y.; Verdin, E.M.; Hsiao, E.C.; Nissenson, R.A. Sirtuin-3 Promotes Adipogenesis, Osteoclastogenesis, and Bone Loss in Aging Male Mice. Endocrinology 2017, 158, 2741–2753. [Google Scholar] [CrossRef]
- AlQranei, M.S.; Senbanjo, L.T.; Aljohani, H.; Hamza, T.; Chellaiah, M.A. Lipopolysaccharide-TLR-4 Axis regulates Osteoclastogenesis independent of RANKL/RANK signaling. BMC Immunol. 2021, 22, 23. [Google Scholar] [CrossRef]
- Oikawa, T.; Oyama, M.; Kozuka-Hata, H.; Uehara, S.; Udagawa, N.; Saya, H.; Matsuo, K. Tks5-dependent formation of circumferential podosomes/invadopodia mediates cell-cell fusion. J. Cell Biol. 2012, 197, 553–568. [Google Scholar] [CrossRef] [Green Version]
- Andela, V.B.; Gordon, A.H.; Zotalis, G.; Rosier, R.N.; Goater, J.J.; Lewis, G.D.; Schwarz, E.M.; Puzas, J.E.; O’Keefe, R.J. NFkappaB: A pivotal transcription factor in prostate cancer metastasis to bone. Clin. Orthop. Relat. Res. 2003, 415, S75–S85. [Google Scholar] [CrossRef]
- Mercatali, L.; Spadazzi, C.; Miserocchi, G.; Liverani, C.; De Vita, A.; Bongiovanni, A.; Recine, F.; Amadori, D.; Ibrahim, T. The Effect of Everolimus in an In Vitro Model of Triple Negative Breast Cancer and Osteoclasts. Int. J. Mol. Sci. 2016, 17, 1827. [Google Scholar] [CrossRef] [Green Version]
- Salamanna, F.; Martini, L.; Pagani, S.; Parrilli, A.; Giavaresi, G.; Maltarello, M.C.; Fini, M. MRMT-1 rat breast carcinoma cells and models of bone metastases: Improvement of an in vitro system to mimic the in vivo condition. Acta Histochem. 2013, 115, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, H.; He, J.; Yuan, X.; Sun, W. Rictor ablation in BMSCs inhibits bone metastasis of TM40D cells by attenuating osteolytic destruction and CAF formation. Int. J. Biol. Sci. 2019, 15, 2448–2460. [Google Scholar] [CrossRef] [PubMed]
- Seol, J.W.; Lee, H.B.; Kim, N.S.; Park, S.Y. Tartrate-resistant acid phosphatase as a diagnostic factor for arthritis. Int. J. Mol. Med. 2009, 24, 57–62. [Google Scholar] [PubMed]
- Esposito, M.; Mondal, N.; Greco, T.M.; Wei, Y.; Spadazzi, C.; Lin, S.C.; Zheng, H.; Cheung, C.; Magnani, J.L.; Lin, S.H.; et al. Bone vascular niche E-selectin induces mesenchymal–epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 2019, 21, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Alshareeda, A.T.; Rakha, E.; Alghwainem, A.; Alrfaei, B.; Alsowayan, B.; Albugami, A.; Alsubayyil, A.M.; Abomraee, M.; Mohd Zin, N.K. The effect of human placental chorionic villi derived mesenchymal stem cell on triple-negative breast cancer hallmarks. PLoS ONE 2018, 13, e0207593. [Google Scholar] [CrossRef]
- Franses, J.W.; Edelman, E.R. The evolution of endothelial regulatory paradigms in cancer biology and vascular repair. Cancer Res. 2011, 71, 7339–7344. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J.; Merler, E.; Abernathy, C.; Williams, G. Isolation of a tumor factor responsible for angiogenesis. J. Exp. Med. 1971, 133, 275–288. [Google Scholar] [CrossRef] [Green Version]
- Van Beijnum, J.R.; Dings, R.P.; van der Linden, E.; Zwaans, B.M.; Ramaekers, F.C.; Mayo, K.H.; Griffioen, A.W. Gene expres-sion of tumor angiogenesis dissected: Specific targeting of colon cancer angiogenic vasculature. Blood 2006, 108, 2339–2348. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, M.; Naczki, C.; Chen, W.; Barlow, K.D.; Case, L.D.; Metheny-Barlow, L.J. Tumor-induced loss of mural Con-nexin 43 gap junction activity promotes endothelial proliferation. BMC Cancer 2015, 15, 427. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chitu, V.; Stanley, E.R.; Elliott, B.E.; Greer, P.A. Fes tyrosine kinase expression in the tumor niche correlates with enhanced tumor growth, angiogenesis, circulating tumor cells, metastasis, and infiltrating macrophages. Cancer Res. 2011, 71, 1465–1473. [Google Scholar] [CrossRef] [Green Version]
- Furlan, A.; Vercamer, C.; Heliot, L.; Wernert, N.; Desbiens, X.; Pourtier, A. Ets-1 drives breast cancer cell angiogenic potential and interactions between breast cancer and endothelial cells. Int. J. Oncol. 2019, 54, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seandel, M.; Butler, J.M.; Kobayashi, H.; Hooper, A.T.; White, I.A.; Zhang, F.; Vertes, E.L.; Kobayashi, M.; Zhang, Y.; Shmelkov, S.V.; et al. Generation of a functional and durable vascular niche by the adenoviral E4ORF1 gene. Proc. Natl. Acad. Sci. USA 2008, 105, 19288–19293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, C.F.; Szot, C.S.; Wilson, T.D.; Akman, S.; Metheny-Barlow, L.J.; Robertson, J.L.; Freeman, J.W.; Rylander, M.N. Cross-talk between endothelial and breast cancer cells regulates reciprocal expression of angiogenic factors in vitro. J. Cell. Biochem. 2012, 113, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, V.; Terzuoli, E.; Donnini, S.; Giachetti, A.; Morbidelli, L.; Ziche, M. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1alpha/VEGF signalling in MCF-7 breast cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 311. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, C.; Viola, M.; Barbouri, D.; Kletsas, D.; Passi, A.; Karamanos, N.K. Dynamic interplay between breast cancer cells and normal endothelium mediates the expression of matrix macromolecules, proteasome activity and functional properties of endothelial cells. Biochim. Biophys. Acta 2014, 1840, 2549–2559. [Google Scholar] [CrossRef] [Green Version]
- Khodarev, N.N.; Yu, J.; Labay, E.; Darga, T.; Brown, C.K.; Mauceri, H.J.; Yassari, R.; Gupta, N.; Weichselbaum, R.R. Tu-mour-endothelium interactions in co-culture: Coordinated changes of gene expression profiles and phenotypic properties of endothelial cells. J. Cell Sci. 2003, 116 (Pt 6), 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Franses, J.W.; Drosu, N.C.; Gibson, W.J.; Chitalia, V.C.; Edelman, E.R. Dysfunctional endothelial cells directly stimulate cancer inflammation and metastasis. Int. J. Cancer 2013, 133, 1334–1344. [Google Scholar] [CrossRef] [Green Version]
- Franses, J.W.; Baker, A.B.; Chitalia, V.C.; Edelman, E.R. Stromal endothelial cells directly influence cancer progression. Sci. Transl. Med. 2011, 3, 66ra5. [Google Scholar] [CrossRef] [Green Version]
- Abraham, V.; Cao, G.; Parambath, A.; Lawal, F.; Handumrongkul, C.; Debs, R.; DeLisser, H.M. Involvement of TIMP-1 in PECAM-1-mediated tumor dissemination. Int. J. Oncol. 2018, 53, 488–502. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.S.; Nam, S.M.; Song, H.K.; Lee, S.; Kim, K.; Lim, H.K.; Lee, H.; Kang, K.T.; Kwon, Y.J.; Chun, Y.J.; et al. CCL8 mediates crosstalk between endothelial colony forming cells and triple-negative breast cancer cells through IL-8, aggravating invasion and tumorigenicity. Oncogene 2021, 40, 3245–3259. [Google Scholar] [CrossRef] [PubMed]
- Ingthorsson, S.; Sigurdsson, V.; Fridriksdottir, A., Jr.; Jonasson, J.G.; Kjartansson, J.; Magnusson, M.K.; Gudjonsson, T. Endo-thelial cells stimulate growth of normal and cancerous breast epithelial cells in 3D culture. BMC Res. Notes 2010, 3, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisdom, K.; Chaudhuri, O. 3D Cell Culture in Interpenetrating Networks of Alginate and rBM Matrix. Methods Mol. Biol. 2017, 1612, 29–37. [Google Scholar] [PubMed]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Rafii, S.; Rafii, A. Endothelial cells provide a notch-dependent pro-tumoral niche for enhancing breast cancer survival, stemness and pro-metastatic properties. PLoS ONE 2014, 9, e112424. [Google Scholar]
- Steinhaeuser, S.S.; Morera, E.; Budkova, Z.; Schepsky, A.; Wang, Q.; Rolfsson, O.; Riedel, A.; Krueger, A.; Hilmarsdottir, B.; Maelandsmo, G.M.; et al. ECM1 secreted by HER2-overexpressing breast cancer cells promotes formation of a vascular niche accelerating cancer cell migration and invasion. Lab. Investig. 2020, 100, 928–944. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J.; Clarke, N.W.; George, N.J.; Shanks, J.H.; Testa, N.G.; Lang, S.H. Interactions of human prostatic epithelial cells with bone marrow endothelium: Binding and invasion. Br. J. Cancer 2001, 84, 1417–1423. [Google Scholar] [CrossRef] [Green Version]
- Connor, Y.; Tekleab, Y.; Tekleab, S.; Nandakumar, S.; Bharat, D.; Sengupta, S. A mathematical model of tumor-endothelial interactions in a 3D co-culture. Sci. Rep. 2019, 9, 8429. [Google Scholar] [CrossRef] [Green Version]
- Shekhar, M.P.; Nangia-Makker, P.; Tait, L.; Miller, F.; Raz, A. Alterations in galectin-3 expression and distribution correlate with breast cancer progression: Functional analysis of galectin-3 in breast epithelial-endothelial interactions. Am. J. Pathol. 2004, 165, 1931–1941. [Google Scholar] [CrossRef]
- Nolan, D.J.; Ginsberg, M.; Israely, E.; Palikuqi, B.; Poulos, M.G.; James, D.; Ding, B.S.; Schachterle, W.; Liu, Y.; Rosenwaks, Z.; et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 2013, 26, 204–219. [Google Scholar] [CrossRef] [Green Version]
- Abraham, E.; Gadish, O.; Franses, J.W.; Chitalia, V.C.; Artzi, N.; Edelman, E.R. Matrix-Embedded Endothelial Cells Attain a Progenitor-Like Phenotype. Adv. Biosyst. 2017, 1, 9. [Google Scholar] [CrossRef]
- Hirschi, K.K.; Ingram, D.A.; Yoder, M.C. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1584–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoder, M.C.; Mead, L.E.; Prater, D.; Krier, T.R.; Mroueh, K.N.; Li, F.; Krasich, R.; Temm, C.J.; Prchal, J.T.; Ingram, D.A. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 2007, 109, 1801–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhode, E.; Malischnik, C.; Thaler, D.; Maierhofer, T.; Linkesch, W.; Lanzer, G.; Guelly, C.; Strunk, D. Blood monocytes mimic endothelial progenitor cells. Stem Cells 2006, 24, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Ronnov-Jessen, L.; Petersen, O.W. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Investig. 1993, 68, 696–707. [Google Scholar] [PubMed]
- Sigurdsson, V.; Fridriksdottir, A.J.; Kjartansson, J.; Jonasson, J.G.; Steinarsdottir, M.; Petersen, O.W.; Ogmundsdottir, H.M.; Gudjonsson, T. Human breast microvascular endothelial cells retain phenotypic traits in long-term finite life span culture. Vitr. Cell. Dev. Biol. Anim. 2006, 42, 332–340. [Google Scholar] [CrossRef]
- Van der Schaft, D.W.; Toebes, E.A.; Haseman, J.R.; Mayo, K.H.; Griffioen, A.W. Bactericidal/permeability-increasing protein (BPI) inhibits angiogenesis via induction of apoptosis in vascular endothelial cells. Blood 2000, 96, 176–181. [Google Scholar] [CrossRef]
- Rizki, A.; Weaver, V.M.; Lee, S.-Y.; Rozenberg, G.I.; Chin, K.; Myers, C.A.; Bascom, J.L.; Mott, J.D.; Semeiks, J.R.; Grate, L.R.; et al. A Human Breast Cell Model of Preinvasive to Invasive Transition. Cancer Res. 2008, 68, 1378–1387. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Nowicka, A.; Solley, T.N.; Wei, C.; Parikh, A.; Court, L.; Burks, J.K.; Andreeff, M.; Woodward, W.A.; Dadbin, A.; et al. Stromal Cells Derived from Visceral and Obese Adipose Tissue Promote Growth of Ovarian Cancers. PLoS ONE 2015, 10, e0136361. [Google Scholar]
- Gao, L.; Zhang, W.; Zhong, W.Q.; Liu, Z.J.; Li, H.M.; Yu, Z.L.; Zhao, Y.F. Tumor associated macrophages induce epithelial to mesenchymal transition via the EGFR/ERK1/2 pathway in head and neck squamous cell carcinoma. Oncol. Rep. 2018, 40, 2558–2572. [Google Scholar] [CrossRef]
- Walker, N.D.; Elias, M.; Guiro, K.; Bhatia, R.; Greco, S.J.; Bryan, M.; Gergues, M.; Sandiford, O.A.; Ponzio, N.M.; Leibovich, S.J.; et al. Exosomes from differentially activated macrophages influence dormancy or resurgence of breast cancer cells within bone marrow stroma. Cell Death Dis. 2019, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.W.; Sceneay, J.; Lima, L.G.; Wong, C.S.; Becker, M.; Krumeich, S.; Lobb, R.J.; Castillo, V.; Wong, K.N.; Ellis, S.; et al. The biodistribution and immune suppressive effects of breast cancer-derived exosomes. Cancer Res. 2016, 76, 6816–6827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, A.L.; Guimaraes, J.C.; Auf der Maur, P.; De Silva, D.; Trefny, M.P.; Okamoto, R.; Bruno, S.; Schmidt, A.; Mertz, K.; Volkmann, K.; et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 2021, 594, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Guereno, M.; Delgado Pastore, M.; Lugones, A.C.; Cercato, M.; Todaro, L.; Urtreger, A.; Peters, M.G. Glypican-3 (GPC3) inhibits metastasis development promoting dormancy in breast cancer cells by p38 MAPK pathway activation. Eur. J. Cell Biol. 2020, 99, 151096. [Google Scholar] [CrossRef]
- Hassan, N.; Greve, B.; Espinoza-Sanchez, N.A.; Gotte, M. Cell-surface heparan sulfate proteoglycans as multifunctional integrators of signaling in cancer. Cell. Signal. 2021, 77, 109822. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Ackerstaff, E.; Artemov, D.; Gillies, R.J.; Bhujwalla, Z.M. Hypoxia and the presence of human vascular endothelial cells affect prostate cancer cell invasion and metabolism. Neoplasia 2007, 9, 1138–1151. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.Q.; Kiefl, R.; Roskopf, C.; Tian, F.; Huber, R.M. Interactions among Lung Cancer Cells, Fibroblasts, and Macrophages in 3D Co-Cultures and the Impact on MMP-1 and VEGF Expression. PLoS ONE 2016, 11, e0156268. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wieder, R. Stromal Co-Cultivation for Modeling Breast Cancer Dormancy in the Bone Marrow. Cancers 2022, 14, 3344. https://doi.org/10.3390/cancers14143344
Wieder R. Stromal Co-Cultivation for Modeling Breast Cancer Dormancy in the Bone Marrow. Cancers. 2022; 14(14):3344. https://doi.org/10.3390/cancers14143344
Chicago/Turabian StyleWieder, Robert. 2022. "Stromal Co-Cultivation for Modeling Breast Cancer Dormancy in the Bone Marrow" Cancers 14, no. 14: 3344. https://doi.org/10.3390/cancers14143344
APA StyleWieder, R. (2022). Stromal Co-Cultivation for Modeling Breast Cancer Dormancy in the Bone Marrow. Cancers, 14(14), 3344. https://doi.org/10.3390/cancers14143344