
“FLipping” the Story: FLT3-Mutated Acute Myeloid Leukemia and the Evolving Role of FLT3 Inhibitors


Abstract
:Simple Summary
Abstract
1. Introduction
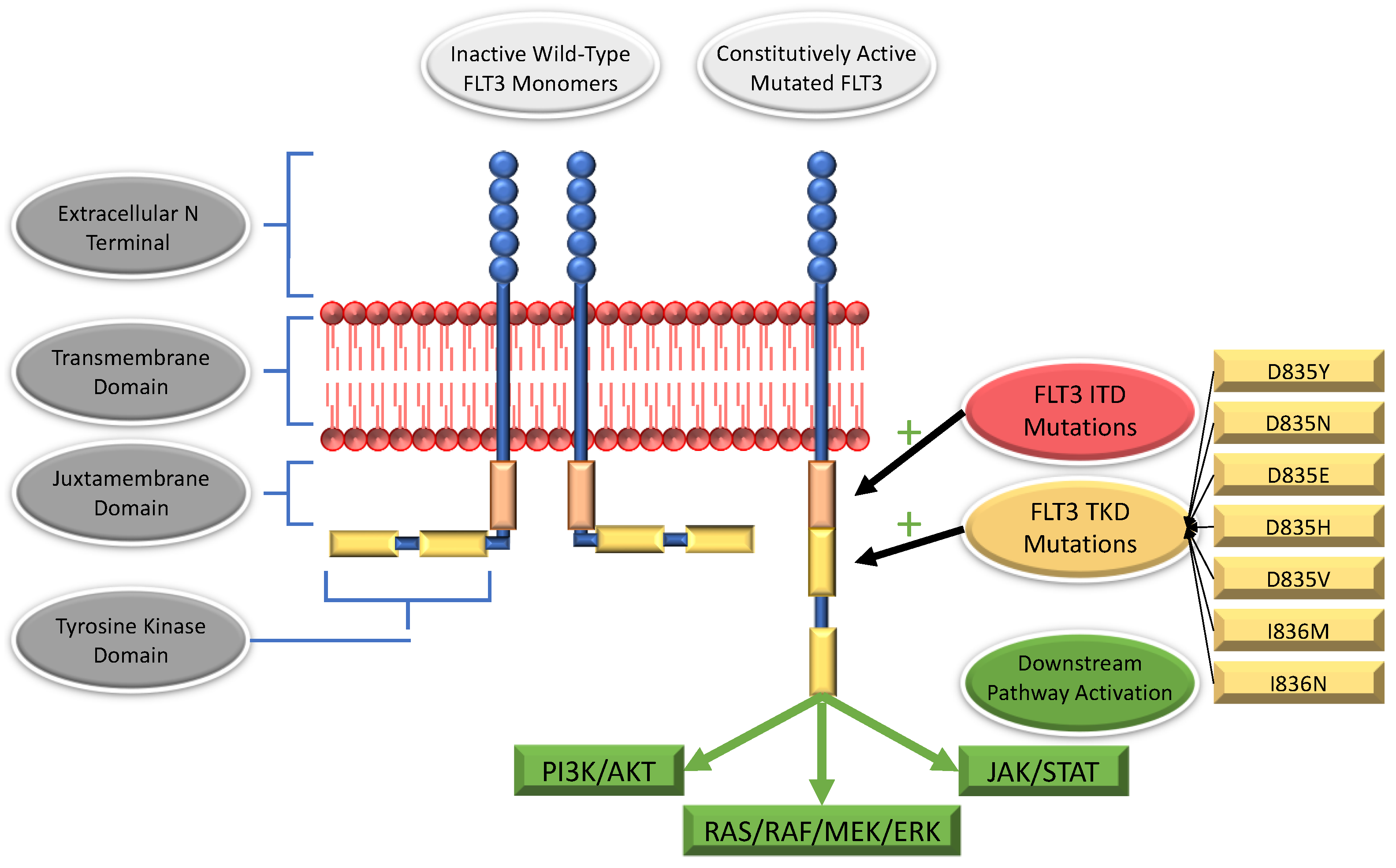
2. The FLT3 Receptor
3. FLT3 Mutations
3.1. Internal Tandem Duplications
3.2. Mutations within the Tyrosine Kinase Domain
3.3. Non-ITD, Non-TKD FLT3 Mutations
3.4. All ITD Mutations Are not Created Equal
3.4.1. Allelic Ratio
3.4.2. Mutation Length
3.4.3. Co-Occurring Mutations
3.5. Implications of FLT3 Mutations for AML Risk Categorization
4. Clinical Implementation of FLT3 Inhibitors in Adult Clinical Trials
4.1. First Generation, Type 1 FLT3 Inhibitors
4.1.1. Midostaurin
4.1.2. Lestaurtinib
4.1.3. Sunitinib
4.2. First Generation, Type 2 FLT3 Inhibitors
4.2.1. Sorafenib
4.2.2. Pexidartinib
4.2.3. Ponatinib
4.3. Second Generation, Type 1 FLT3 Inhibitors
4.3.1. Gilteritinib
4.3.2. Crenolanib
4.3.3. MRX-2843
4.4. Second Generation, Type 2 FLT3 Inhibitors
Quizartinib
5. FLT3 Inhibition in Pediatric FLT3-Mutated AML
6. CD33+ Targeting, Consolidative HSCT, and Integration of FLT3 Inhibition
7. Resistance to FLT3 Inhibition—And How to Overcome It
7.1. Intrinsic Mechanisms of Resistance
7.2. Emergent Mechanisms of Resistance
8. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute Myeloid Leukemia: Current Progress and Future Directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Appelbaum, F.R.; Gundacker, H.; Head, D.R.; Slovak, M.L.; Willman, C.L.; Godwin, J.E.; Anderson, J.E.; Petersdorf, S.H. Age and Acute Myeloid Leukemia. Blood 2006, 107, 3481–3485. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; et al. Genomic and Epigenomic Landscapes of Adult de Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, M.; Yamaguchi, H.; Kuboyama, M.; Najima, Y.; Usuki, K.; Ueki, T.; Oh, I.; Mori, S.; Kawata, E.; Uoshima, N.; et al. Significance of FLT3-Tyrosine Kinase Domain Mutation as a Prognostic Factor for Acute Myeloid Leukemia. Int. J. Hematol. 2019, 110, 566–574. [Google Scholar] [CrossRef]
- Bacher, U.; Haferlach, C.; Kern, W.; Haferlach, T.; Schnittger, S. Prognostic Relevance of FLT3-TKD Mutations in AML: The Combination Matters—An Analysis of 3082 Patients. Blood 2008, 111, 2527–2537. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 Mutations in AML: Review of Current Knowledge and Evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Kottaridis, P.D.; Gale, R.E.; Frew, M.E.; Harrison, G.; Langabeer, S.E.; Belton, A.A.; Walker, H.; Wheatley, K.; Bowen, D.T.; Burnett, A.K.; et al. The Presence of a FLT3 Internal Tandem Duplication in Patients with Acute Myeloid Leukemia (AML) Adds Important Prognostic Information to Cytogenetic Risk Group and Response to the First Cycle of Chemotherapy: Analysis of 854 Patients from the United Kingdom Medical Research Council AML 10 and 12 Trials. Blood 2001, 98, 1752–1759. [Google Scholar] [CrossRef]
- Kiyoi, H.; Naoe, T.; Nakano, Y.; Yokota, S.; Minami, S.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Jinnai, I.; Shimazaki, C.; et al. Prognostic Implication of FLT3 and N-RAS Gene Mutations in Acute Myeloid Leukemia. Blood 1999, 93, 3074–3080. [Google Scholar]
- Port, M.; Böttcher, M.; Thol, F.; Ganser, A.; Schlenk, R.; Wasem, J.; Neumann, A.; Pouryamout, L. Prognostic Significance of FLT3 Internal Tandem Duplication, Nucleophosmin 1, and CEBPA Gene Mutations for Acute Myeloid Leukemia Patients with Normal Karyotype and Younger than 60 Years: A Systematic Review and Meta-Analysis. Ann. Hematol. 2014, 93, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, A.; Assouline, S. A Review of FDA-Approved Acute Myeloid Leukemia Therapies beyond “7 + 3”. Expert Rev. Hematol. 2021, 14, 185–197. [Google Scholar] [CrossRef]
- Takahashi, S. Downstream Molecular Pathways of FLT3 in the Pathogenesis of Acute Myeloid Leukemia: Biology and Therapeutic Implications. J. Hematol. Oncol. J. Hematol. Oncol. 2011, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levis, M.; Small, D. FLT3: ITDoes Matter in Leukemia. Leukemia 2003, 17, 1738–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenstein, R. Class III Receptor Tyrosine Kinases in Acute Leukemia—Biological Functions and Modern Laboratory Analysis. Biomark. Insights 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todde, G.; Friedman, R. Activation and Inactivation of the FLT3 Kinase: Pathway Intermediates and the Free Energy of Transition. J. Phys. Chem. B 2019, 123, 5385–5394. [Google Scholar] [CrossRef]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An Overview on the Role of FLT3-Tyrosine Kinase Receptor in Acute Myeloid Leukemia: Biology and Treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef] [Green Version]
- Nitika; Wei, J.; Hui, A.-M. Role of Biomarkers in FLT3 AML. Cancers 2022, 14, 1164. [Google Scholar] [CrossRef]
- Kazi, J.U.; Rönnstrand, L. FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef]
- Matthews, W.; Jordan, C.T.; Wiegand, G.W.; Pardoll, D.; Lemischka, I.R. A Receptor Tyrosine Kinase Specific to Hematopoietic Stem and Progenitor Cell-Enriched Populations. Cell 1991, 65, 1143–1152. [Google Scholar] [CrossRef]
- Brasel, K.; Escobar, S.; Anderberg, R.; de Vries, P.; Gruss, H.J.; Lyman, S.D. Expression of the Flt3 Receptor and Its Ligand on Hematopoietic Cells. Leukemia 1995, 9, 1212–1218. [Google Scholar] [PubMed]
- Rosnet, O.; Bühring, H.J.; Marchetto, S.; Rappold, I.; Lavagna, C.; Sainty, D.; Arnoulet, C.; Chabannon, C.; Kanz, L.; Hannum, C.; et al. Human FLT3/FLK2 Receptor Tyrosine Kinase Is Expressed at the Surface of Normal and Malignant Hematopoietic Cells. Leukemia 1996, 10, 238–248. [Google Scholar] [PubMed]
- Tissue Expression of FLT3—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000122025-FLT3/tissue (accessed on 12 May 2022).
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell. Proteom. MCP 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Kikushige, Y.; Yoshimoto, G.; Miyamoto, T.; Iino, T.; Mori, Y.; Iwasaki, H.; Niiro, H.; Takenaka, K.; Nagafuji, K.; Harada, M.; et al. Human Flt3 Is Expressed at the Hematopoietic Stem Cell and the Granulocyte/Macrophage Progenitor Stages to Maintain Cell Survival. J. Immunol. Baltim. Md 1950 2008, 180, 7358–7367. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; McPherson, C.M.; Stambrook, P.J. Flt-3 Ligand: A Potent Dendritic Cell Stimulator and Novel Antitumor Agent. Cancer Biol. Ther. 2002, 1, 486–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eidenschenk, C.; Crozat, K.; Krebs, P.; Arens, R.; Popkin, D.; Arnold, C.N.; Blasius, A.L.; Benedict, C.A.; Moresco, E.M.Y.; Xia, Y.; et al. Flt3 Permits Survival during Infection by Rendering Dendritic Cells Competent to Activate NK Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 9759–9764. [Google Scholar] [CrossRef] [Green Version]
- Meshinchi, S.; Alonzo, T.A.; Stirewalt, D.L.; Zwaan, M.; Zimmerman, M.; Reinhardt, D.; Kaspers, G.J.L.; Heerema, N.A.; Gerbing, R.; Lange, B.J.; et al. Clinical Implications of FLT3 Mutations in Pediatric AML. Blood 2006, 108, 3654–3661. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef]
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal Tandem Duplication of the Flt3 Gene Found in Acute Myeloid Leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar]
- Zhang, S.; Broxmeyer, H.E. Flt3 Ligand Induces Tyrosine Phosphorylation of Gab1 and Gab2 and Their Association with Shp-2, Grb2, and PI3 Kinase. Biochem. Biophys. Res. Commun. 2000, 277, 195–199. [Google Scholar] [CrossRef]
- Chen, P.; Levis, M.; Brown, P.; Kim, K.-T.; Allebach, J.; Small, D. FLT3/ITD Mutation Signaling Includes Suppression of SHP-1. J. Biol. Chem. 2005, 280, 5361–5369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, R.; Friedman, A.D.; Levis, M.; Li, L.; Weir, E.G.; Small, D. Internal Tandem Duplication Mutation of FLT3 Blocks Myeloid Differentiation through Suppression of C/EBPalpha Expression. Blood 2004, 103, 1883–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tickenbrock, L.; Schwäble, J.; Wiedehage, M.; Steffen, B.; Sargin, B.; Choudhary, C.; Brandts, C.; Berdel, W.E.; Müller-Tidow, C.; Serve, H. Flt3 Tandem Duplication Mutations Cooperate with Wnt Signaling in Leukemic Signal Transduction. Blood 2005, 105, 3699–3706. [Google Scholar] [CrossRef]
- Hayakawa, F.; Towatari, M.; Kiyoi, H.; Tanimoto, M.; Kitamura, T.; Saito, H.; Naoe, T. Tandem-Duplicated Flt3 Constitutively Activates STAT5 and MAP Kinase and Introduces Autonomous Cell Growth in IL-3-Dependent Cell Lines. Oncogene 2000, 19, 624–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitenbuecher, F.; Schnittger, S.; Grundler, R.; Markova, B.; Carius, B.; Brecht, A.; Duyster, J.; Haferlach, T.; Huber, C.; Fischer, T. Identification of a Novel Type of ITD Mutations Located in Nonjuxtamembrane Domains of the FLT3 Tyrosine Kinase Receptor. Blood 2009, 113, 4074–4077. [Google Scholar] [CrossRef]
- Rücker, F.G.; Du, L.; Luck, T.J.; Benner, A.; Krzykalla, J.; Gathmann, I.; Voso, M.T.; Amadori, S.; Prior, T.W.; Brandwein, J.M.; et al. Molecular Landscape and Prognostic Impact of FLT3-ITD Insertion Site in Acute Myeloid Leukemia: RATIFY Study Results. Leukemia 2022, 36, 90–99. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kiyoi, H.; Nakano, Y.; Suzuki, R.; Kodera, Y.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Yagasaki, F.; Shimazaki, C.; et al. Activating Mutation of D835 within the Activation Loop of FLT3 in Human Hematologic Malignancies. Blood 2001, 97, 2434–2439. [Google Scholar] [CrossRef] [Green Version]
- Scholl, S.; Krause, C.; Loncarevic, I.F.; Müller, R.; Kunert, C.; Wedding, U.; Sayer, H.G.; Clement, J.H.; Höffken, K. Specific Detection of Flt3 Point Mutations by Highly Sensitive Real-Time Polymerase Chain Reaction in Acute Myeloid Leukemia. J. Lab. Clin. Med. 2005, 145, 295–304. [Google Scholar] [CrossRef]
- Choudhary, C.; Schwäble, J.; Brandts, C.; Tickenbrock, L.; Sargin, B.; Kindler, T.; Fischer, T.; Berdel, W.E.; Müller-Tidow, C.; Serve, H. AML-Associated Flt3 Kinase Domain Mutations Show Signal Transduction Differences Compared with Flt3 ITD Mutations. Blood 2005, 106, 265–273. [Google Scholar] [CrossRef]
- Mead, A.J.; Linch, D.C.; Hills, R.K.; Wheatley, K.; Burnett, A.K.; Gale, R.E. FLT3 Tyrosine Kinase Domain Mutations Are Biologically Distinct from and Have a Significantly More Favorable Prognosis than FLT3 Internal Tandem Duplications in Patients with Acute Myeloid Leukemia. Blood 2007, 110, 1262–1270. [Google Scholar] [CrossRef]
- Staffas, A.; Kanduri, M.; Hovland, R.; Rosenquist, R.; Ommen, H.B.; Abrahamsson, J.; Forestier, E.; Jahnukainen, K.; Jónsson, Ó.G.; Zeller, B.; et al. Presence of FLT3-ITD and High BAALC Expression Are Independent Prognostic Markers in Childhood Acute Myeloid Leukemia. Blood 2011, 118, 5905–5913. [Google Scholar] [CrossRef]
- Young, D.J.; Nguyen, B.; Zhu, R.; Seo, J.; Li, L.; Levis, M.J.; Pratz, K.W.; Duffield, A.S.; Small, D. Deletions in FLT-3 Juxtamembrane Domain Define a New Class of Pathogenic Mutations: Case Report and Systematic Analysis. Blood Adv. 2021, 5, 2285–2293. [Google Scholar] [CrossRef]
- Cosmic COSMIC—Catalogue of Somatic Mutations in Cancer. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 12 May 2022).
- Meshinchi, S.; Stirewalt, D.L.; Alonzo, T.A.; Boggon, T.J.; Gerbing, R.B.; Rocnik, J.L.; Lange, B.J.; Gilliland, D.G.; Radich, J.P. Structural and Numerical Variation of FLT3/ITD in Pediatric AML. Blood 2008, 111, 4930–4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarlock, K.; Hansen, M.E.; Hylkema, T.; Ries, R.; Farrar, J.E.; Auvil, J.G.; Gerhard, D.S.; Smith, M.A.; Davidsen, T.M.; Gesuwan, P.; et al. Discovery and Functional Validation of Novel Pediatric Specific FLT3 Activating Mutations in Acute Myeloid Leukemia: Results from the COG/NCI Target Initiative. Blood 2015, 126, 87. [Google Scholar] [CrossRef]
- Levis, M. FLT3 Mutations in Acute Myeloid Leukemia: What Is the Best Approach in 2013? Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 220–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, C.; Linch, D.C.; Gale, R.E. Most Acute Myeloid Leukaemia Patients with Intermediate Mutant FLT3/ITD Levels Do Not Have Detectable Bi-Allelic Disease, Indicating That Heterozygous Disease Alone Is Associated with an Adverse Outcome. Br. J. Haematol. 2008, 142, 423–426. [Google Scholar] [CrossRef]
- Whitman, S.P.; Archer, K.J.; Feng, L.; Baldus, C.; Becknell, B.; Carlson, B.D.; Carroll, A.J.; Mrózek, K.; Vardiman, J.W.; George, S.L.; et al. Absence of the Wild-Type Allele Predicts Poor Prognosis in Adult de Novo Acute Myeloid Leukemia with Normal Cytogenetics and the Internal Tandem Duplication of FLT3: A Cancer and Leukemia Group B Study. Cancer Res. 2001, 61, 7233–7239. [Google Scholar] [PubMed]
- Blau, O.; Berenstein, R.; Sindram, A.; Blau, I.W. Molecular Analysis of Different FLT3-ITD Mutations in Acute Myeloid Leukemia. Leuk. Lymphoma 2013, 54, 145–152. [Google Scholar] [CrossRef]
- Linch, D.C.; Hills, R.K.; Burnett, A.K.; Khwaja, A.; Gale, R.E. Impact of FLT3(ITD) Mutant Allele Level on Relapse Risk in Intermediate-Risk Acute Myeloid Leukemia. Blood 2014, 124, 273–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, G.; Chopra, Y.; Mourad, S.; Chiang, K.-Y.; Hitzler, J. Treatment of Acute Myeloid Leukemia in Children: A Practical Perspective. Pediatr. Blood Cancer 2021, 68, e28979. [Google Scholar] [CrossRef]
- Abou Dalle, I.; Ghorab, A.; Patel, K.; Wang, X.; Hwang, H.; Cortes, J.; Issa, G.C.; Yalniz, F.; Sasaki, K.; Chihara, D.; et al. Impact of Numerical Variation, Allele Burden, Mutation Length and Co-Occurring Mutations on the Efficacy of Tyrosine Kinase Inhibitors in Newly Diagnosed FLT3- Mutant Acute Myeloid Leukemia. Blood Cancer J. 2020, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Döhner, K.; Thiede, C.; Jahn, N.; Panina, E.; Gambietz, A.; Larson, R.A.; Prior, T.W.; Marcucci, G.; Jones, D.; Krauter, J.; et al. Impact of NPM1/FLT3-ITD Genotypes Defined by the 2017 European LeukemiaNet in Patients with Acute Myeloid Leukemia. Blood 2020, 135, 371–380. [Google Scholar] [CrossRef]
- Tarlock, K.; Zhong, S.; He, Y.; Ries, R.; Severson, E.; Bailey, M.; Morley, S.; Balasubramanian, S.; Erlich, R.; Lipson, D.; et al. Distinct Age-Associated Molecular Profiles in Acute Myeloid Leukemia Defined by Comprehensive Clinical Genomic Profiling. Oncotarget 2018, 9, 26417–26430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schäkel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-Activating Mutations in 979 Patients with Acute Myelogenous Leukemia: Association with FAB Subtypes and Identification of Subgroups with Poor Prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef] [Green Version]
- Zwaan, C.M.; Meshinchi, S.; Radich, J.P.; Veerman, A.J.P.; Huismans, D.R.; Munske, L.; Podleschny, M.; Hählen, K.; Pieters, R.; Zimmermann, M.; et al. FLT3 Internal Tandem Duplication in 234 Children with Acute Myeloid Leukemia: Prognostic Significance and Relation to Cellular Drug Resistance. Blood 2003, 102, 2387–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.B.; Heerema-McKenney, A.; Winter, L.; et al. Gemtuzumab Ozogamicin in Children and Adolescents with De Novo Acute Myeloid Leukemia Improves Event-Free Survival by Reducing Relapse Risk: Results From the Randomized Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2014, 32, 3021–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarlock, K.; Alonzo, T.A.; Gerbing, R.B.; Ries, R.E.; Gibson, B.; Niktoreh, N.; Noort, S.; Van den Heuvel-Eibrink, M.M.; Zwaan, C.M.; Meshinchi, S. Distinct Co-Occurring Mutational Profiles in Acute Myeloid Leukemia Confers Prognostic Significance in Children and Young Adults with FLT3/ITD Mutations. Blood 2018, 132, 443. [Google Scholar] [CrossRef]
- Children’s Oncology Group. A Phase 3 Randomized Trial for Patients with De Novo AML Comparing Standard Therapy Including Gemtuzumab Ozogamicin (GO) to CPX-351 with GO, and the Addition of the FLT3 Inhibitor Gilteritinib for Patients with FLT3 Mutations; Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- Koszarska, M.; Meggyesi, N.; Bors, A.; Batai, A.; Csacsovszki, O.; Lehoczky, E.; Adam, E.; Kozma, A.; Lovas, N.; Sipos, A.; et al. Medium-Sized FLT3 Internal Tandem Duplications Confer Worse Prognosis than Short and Long Duplications in a Non-Elderly Acute Myeloid Leukemia Cohort. Leuk. Lymphoma 2014, 55, 1510–1517. [Google Scholar] [CrossRef]
- Castaño-Bonilla, T.; Alonso-Dominguez, J.M.; Barragán, E.; Rodríguez-Veiga, R.; Sargas, C.; Gil, C.; Chillón, C.; Vidriales, M.B.; García, R.; Martínez-López, J.; et al. Prognostic Significance of FLT3-ITD Length in AML Patients Treated with Intensive Regimens. Sci. Rep. 2021, 11, 20745. [Google Scholar] [CrossRef]
- Garg, M.; Nagata, Y.; Kanojia, D.; Mayakonda, A.; Yoshida, K.; Haridas Keloth, S.; Zang, Z.J.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; et al. Profiling of Somatic Mutations in Acute Myeloid Leukemia with FLT3-ITD at Diagnosis and Relapse. Blood 2015, 126, 2491–2501. [Google Scholar] [CrossRef] [Green Version]
- Kirtonia, A.; Pandya, G.; Sethi, G.; Pandey, A.K.; Das, B.C.; Garg, M. A Comprehensive Review of Genetic Alterations and Molecular Targeted Therapies for the Implementation of Personalized Medicine in Acute Myeloid Leukemia. J. Mol. Med. Berl. Ger. 2020, 98, 1069–1091. [Google Scholar] [CrossRef] [PubMed]
- Tarlock, K.; Alonzo, T.A.; Moraleda, P.P.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Ravindranath, Y.; Lange, B.; Woods, W.G.; Gamis, A.S.; et al. Acute Myeloid Leukaemia (AML) with t(6;9)(P23;Q34) Is Associated with Poor Outcome in Childhood AML Regardless of FLT3-ITD Status: A Report from the Children’s Oncology Group. Br. J. Haematol. 2014, 166, 254–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarlock, K. Significant Improvements in Survival for Patients with t(6;9)(P23;Q34)/DEK-NUP214 in Contemporary Trials with Intensification of Therapy: A Report from the Children’s Oncology Group; ASH: Washington, DC, USA, 2021. [Google Scholar]
- Oran, B.; Cortes, J.; Beitinjaneh, A.; Chen, H.-C.; de Lima, M.; Patel, K.; Ravandi, F.; Wang, X.; Brandt, M.; Andersson, B.S.; et al. Allogeneic Transplantation in First Remission Improves Outcomes Irrespective of FLT3-ITD Allelic Ratio in FLT3-ITD-Positive Acute Myelogenous Leukemia. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2016, 22, 1218–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratcorona, M.; Brunet, S.; Nomdedéu, J.; Ribera, J.M.; Tormo, M.; Duarte, R.; Escoda, L.; Guàrdia, R.; Queipo de Llano, M.P.; Salamero, O.; et al. Favorable Outcome of Patients with Acute Myeloid Leukemia Harboring a Low-Allelic Burden FLT3-ITD Mutation and Concomitant NPM1 Mutation: Relevance to Post-Remission Therapy. Blood 2013, 121, 2734–2738. [Google Scholar] [CrossRef] [Green Version]
- Schlenk, R.F.; Kayser, S.; Bullinger, L.; Kobbe, G.; Casper, J.; Ringhoffer, M.; Held, G.; Brossart, P.; Lübbert, M.; Salih, H.R.; et al. Differential Impact of Allelic Ratio and Insertion Site in FLT3-ITD-Positive AML with Respect to Allogeneic Transplantation. Blood 2014, 124, 3441–3449. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.D.; Schetelig, J.; Bochtler, T.; Schaich, M.; Schäfer-Eckart, K.; Hänel, M.; Rösler, W.; Einsele, H.; Kaufmann, M.; Serve, H.; et al. Allogeneic Stem Cell Transplantation Improves Survival in Patients with Acute Myeloid Leukemia Characterized by a High Allelic Ratio of Mutant FLT3-ITD. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2016, 22, 462–469. [Google Scholar] [CrossRef] [Green Version]
- Pratz, K.W.; Levis, M. How I Treat FLT3-Mutated AML. Blood 2017, 129, 565–571. [Google Scholar] [CrossRef]
- Boddu, P.; Kantarjian, H.; Borthakur, G.; Kadia, T.; Daver, N.; Pierce, S.; Andreeff, M.; Ravandi, F.; Cortes, J.; Kornblau, S.M. Co-Occurrence of FLT3-TKD and NPM1 Mutations Defines a Highly Favorable Prognostic AML Group. Blood Adv. 2017, 1, 1546–1550. [Google Scholar] [CrossRef] [Green Version]
- Esnault, C.; Rahmé, R.; Rice, K.L.; Berthier, C.; Gaillard, C.; Quentin, S.; Maubert, A.-L.; Kogan, S.; de Thé, H. FLT3-ITD Impedes Retinoic Acid, but Not Arsenic, Responses in Murine Acute Promyelocytic Leukemias. Blood 2019, 133, 1495–1506. [Google Scholar] [CrossRef]
- Noguera, N.I.; Breccia, M.; Divona, M.; Diverio, D.; Costa, V.; De Santis, S.; Avvisati, G.; Pinazzi, M.B.; Petti, M.C.; Mandelli, F.; et al. Alterations of the FLT3 Gene in Acute Promyelocytic Leukemia: Association with Diagnostic Characteristics and Analysis of Clinical Outcome in Patients Treated with the Italian AIDA Protocol. Leukemia 2002, 16, 2185–2189. [Google Scholar] [CrossRef] [Green Version]
- Picharski, G.L.; Andrade, D.P.; Fabro, A.L.M.R.; Lenzi, L.; Tonin, F.S.; Ribeiro, R.C.; Figueiredo, B.C. The Impact of Flt3 Gene Mutations in Acute Promyelocytic Leukemia: A Meta-Analysis. Cancers 2019, 11, 1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, M.R.; Tallman, M.S.; Abboud, C.N.; Altman, J.K.; Appelbaum, F.R.; Arber, D.A.; Bhatt, V.; Bixby, D.; Blum, W.; Coutre, S.E.; et al. Acute Myeloid Leukemia, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. JNCCN 2017, 15, 926–957. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations from an International Expert Panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Man, C.H.; Fung, T.K.; Ho, C.; Han, H.H.C.; Chow, H.C.H.; Ma, A.C.H.; Choi, W.W.L.; Lok, S.; Cheung, A.M.S.; Eaves, C.; et al. Sorafenib Treatment of FLT3-ITD(+) Acute Myeloid Leukemia: Favorable Initial Outcome and Mechanisms of Subsequent Nonresponsiveness Associated with the Emergence of a D835 Mutation. Blood 2012, 119, 5133–5143. [Google Scholar] [CrossRef]
- Smith, C.C. The Growing Landscape of FLT3 Inhibition in AML. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 539–547. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Voso, M.T.; Larson, R.A.; Jones, D.; Marcucci, G.; Prior, T.; Krauter, J.; Heuser, M.; Lavorgna, S.; Nomdedeu, J.; Geyer, S.M.; et al. Midostaurin in Patients with Acute Myeloid Leukemia and FLT3-TKD Mutations: A Subanalysis from the RATIFY Trial. Blood Adv. 2020, 4, 4945–4954. [Google Scholar] [CrossRef]
- Maziarz, R.T.; Levis, M.; Patnaik, M.M.; Scott, B.L.; Mohan, S.R.; Deol, A.; Rowley, S.D.; Kim, D.D.H.; Hernandez, D.; Rajkhowa, T.; et al. Midostaurin after Allogeneic Stem Cell Transplant in Patients with FLT3-Internal Tandem Duplication-Positive Acute Myeloid Leukemia. Bone Marrow Transplant. 2021, 56, 1180–1189. [Google Scholar] [CrossRef]
- Lai, C.; Doucette, K.; Norsworthy, K. Recent Drug Approvals for Acute Myeloid Leukemia. J. Hematol. Oncol.J Hematol Oncol 2019, 12, 100. [Google Scholar] [CrossRef]
- Knapper, S.; Russell, N.; Gilkes, A.; Hills, R.K.; Gale, R.E.; Cavenagh, J.D.; Jones, G.; Kjeldsen, L.; Grunwald, M.R.; Thomas, I.; et al. A Randomized Assessment of Adding the Kinase Inhibitor Lestaurtinib to First-Line Chemotherapy for FLT3-Mutated AML. Blood 2017, 129, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, W.; Kayser, S.; Kebenko, M.; Janning, M.; Krauter, J.; Schittenhelm, M.; Götze, K.; Weber, D.; Göhring, G.; Teleanu, V.; et al. A Phase I/II Study of Sunitinib and Intensive Chemotherapy in Patients over 60 Years of Age with Acute Myeloid Leukaemia and Activating FLT3 Mutations. Br. J. Haematol. 2015, 169, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Röllig, C.; Serve, H.; Hüttmann, A.; Noppeney, R.; Müller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of Sorafenib versus Placebo to Standard Therapy in Patients Aged 60 Years or Younger with Newly Diagnosed Acute Myeloid Leukaemia (SORAML): A Multicentre, Phase 2, Randomised Controlled Trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Röllig, C.; Serve, H.; Noppeney, R.; Hanoun, M.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; Krämer, A.; et al. Sorafenib or Placebo in Patients with Newly Diagnosed Acute Myeloid Leukaemia: Long-Term Follow-up of the Randomized Controlled SORAML Trial. Leukemia 2021, 35, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Kennedy, G.A.; Morris, K.L.; Grigg, A.; He, S.; Schwarer, A.; Ting, S.B.; Enjeti, A.K.; Yuen, S.; D’Rozario, J.; et al. Results of a Phase 2, Randomized, Double-Blind Study of Sorafenib Versus Placebo in Combination with Intensive Chemotherapy in Previously Untreated Patients with FLT3-ITD Acute Myeloid Leukemia (ALLG AMLM16). Blood 2020, 136, 36–38. [Google Scholar] [CrossRef]
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, J.; Stelljes, M.; Röllig, C.; Wollmer, E.; Wäsch, R.; Bornhäuser, M.; Berg, T.; et al. Sorafenib Maintenance After Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia with FLT3–Internal Tandem Duplication Mutation (SORMAIN). J. Clin. Oncol. 2020, 38, 2993–3002. [Google Scholar] [CrossRef]
- Smith, C.C.; Levis, M.J.; Frankfurt, O.; Pagel, J.M.; Roboz, G.J.; Stone, R.M.; Wang, E.S.; Severson, P.L.; West, B.L.; Le, M.H.; et al. A Phase 1/2 Study of the Oral FLT3 Inhibitor Pexidartinib in Relapsed/Refractory FLT3-ITD-Mutant Acute Myeloid Leukemia. Blood Adv. 2020, 4, 1711–1721. [Google Scholar] [CrossRef]
- Smith, C.C.; Lasater, E.A.; Zhu, X.; Lin, K.C.; Stewart, W.K.; Damon, L.E.; Salerno, S.; Shah, N.P. Activity of Ponatinib against Clinically-Relevant AC220-Resistant Kinase Domain Mutants of FLT3-ITD. Blood 2013, 121, 3165–3171. [Google Scholar] [CrossRef] [Green Version]
- Talpaz, M.; Shah, N.P.; Deininger, M.W.; Mauro, M.J.; Flinn, I.W.; Lustgarten, S.; Lindmark, W.; Gozgit, J.M.; Clackson, T.; Turner, C.D.; et al. Ponatinib in Patients with Acute Myeloid Leukemia (AML): Preliminary Findings from a Phase I Study in Hematologic Malignancies. J. Clin. Oncol. 2011, 29, 6518. [Google Scholar] [CrossRef]
- Shah, N.P.; Talpaz, M.; Deininger, M.W.N.; Mauro, M.J.; Flinn, I.W.; Bixby, D.; Lustgarten, S.; Gozgit, J.M.; Clackson, T.; Turner, C.D.; et al. Ponatinib in Patients with Refractory Acute Myeloid Leukaemia: Findings from a Phase 1 Study. Br. J. Haematol. 2013, 162, 548–552. [Google Scholar] [CrossRef] [Green Version]
- Kipp, D.; Loo, S.; Perkins, A.C.; Lane, S.W.; Blyth, E.; Enjeti, A.K.; Bajel, A.; Reynolds, J.; Wei, A.H. A Phase-Ib/II Clinical Evaluation of Ponatinib in Combination with Azacitidine in FLT3-ITD and CBL-Mutant Acute Myeloid Leukemia (PON-AZA Study). Blood 2021, 138, 2350. [Google Scholar] [CrossRef]
- Lee, L.Y.; Hernandez, D.; Rajkhowa, T.; Smith, S.C.; Raman, J.R.; Nguyen, B.; Small, D.; Levis, M. Preclinical Studies of Gilteritinib, a next-Generation FLT3 Inhibitor. Blood 2017, 129, 257–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, M.; Kaneko, N.; Ueno, Y.; Yamada, M.; Tanaka, R.; Saito, R.; Shimada, I.; Mori, K.; Kuromitsu, S. Gilteritinib, a FLT3/AXL Inhibitor, Shows Antileukemic Activity in Mouse Models of FLT3 Mutated Acute Myeloid Leukemia. Investig. New Drugs 2017, 35, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective Inhibition of FLT3 by Gilteritinib in Relapsed or Refractory Acute Myeloid Leukaemia: A Multicentre, First-in-Human, Open-Label, Phase 1–2 Study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Larson, R.A.; Podoltsev, N.A.; Strickland, S.A.; Wang, E.S.; Atallah, E.; Schiller, G.J.; Martinelli, G.; Neubauer, A.; Sierra, J.; et al. Follow-up of Patients with R/R FLT3-Mutation-Positive AML Treated with Gilteritinib in the Phase 3 ADMIRAL Trial. Blood 2022, 139, 3366–3375. [Google Scholar] [CrossRef]
- Pratz, K. A Phase 1 Study of Gilteritinib in Combination with Induction and Consolidation Chemotherapy in Patients with Newly Diagnosed AML: Final Results; ASH: Washington, DC, USA, 2020. [Google Scholar]
- Cortes, J.E.; Kantarjian, H.M.; Kadia, T.M.; Borthakur, G.; Konopleva, M.; Garcia-Manero, G.; Daver, N.G.; Pemmaraju, N.; Jabbour, E.; Estrov, Z.; et al. Crenolanib Besylate, a Type I Pan-FLT3 Inhibitor, to Demonstrate Clinical Activity in Multiply Relapsed FLT3-ITD and D835 AML. J. Clin. Oncol. 2016, 34, 7008. [Google Scholar] [CrossRef]
- Iyer, S.P.; Jethava, Y.; Karanes, C.; Eckardt, J.R.; Collins, R. Safety Study of Salvage Chemotherapy High-Dose Ara-C/Mitoxantrone (HAM) and Type I FLT3-TKI Crenolanib in First Relapsed/Primary Refractory AML. Blood 2016, 128, 3983. [Google Scholar] [CrossRef]
- Aboudalle, I.; Kantarjian, H.M.; Ohanian, M.N.; Alvarado, Y.; Jabbour, E.J.; Garcia-Manero, G.; Naqvi, K.; Wierda, W.G.; Daver, N.G.; Burger, J.A.; et al. Phase I-II Study of Crenolanib Combined with Standard Salvage Chemotherapy and Crenolanib Combined with 5-Azacitidine in Acute Myeloid Leukemia Patients with FLT3 Activating Mutations. Blood 2018, 132, 2715. [Google Scholar] [CrossRef]
- Wang, E.S.; Tallman, M.S.; Stone, R.M.; Walter, R.B.; Karanes, C.; Jain, V.; Collins, R.H. Low Relapse Rate in Younger Patients ≤ 60 Years Old with Newly Diagnosed FLT3-Mutated Acute Myeloid Leukemia (AML) Treated with Crenolanib and Cytarabine/Anthracycline Chemotherapy. Blood 2017, 130, 566. [Google Scholar] [CrossRef]
- Lee-Sherick, A.B.; Eisenman, K.M.; Sather, S.; McGranahan, A.; Armistead, P.M.; McGary, C.S.; Hunsucker, S.A.; Schlegel, J.; Martinson, H.; Cannon, C.; et al. Aberrant Mer Receptor Tyrosine Kinase Expression Contributes to Leukemogenesis in Acute Myeloid Leukemia. Oncogene 2013, 32, 5359–5368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Sherick, A.B.; Zhang, W.; Menachof, K.K.; Hill, A.A.; Rinella, S.; Kirkpatrick, G.; Page, L.S.; Stashko, M.A.; Jordan, C.T.; Wei, Q.; et al. Efficacy of a Mer and Flt3 Tyrosine Kinase Small Molecule Inhibitor, UNC1666, in Acute Myeloid Leukemia. Oncotarget 2015, 6, 6722–6736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minson, K.A.; Smith, C.C.; DeRyckere, D.; Libbrecht, C.; Lee-Sherick, A.B.; Huey, M.G.; Lasater, E.A.; Kirkpatrick, G.D.; Stashko, M.A.; Zhang, W.; et al. The MERTK/FLT3 Inhibitor MRX-2843 Overcomes Resistance-Conferring FLT3 Mutations in Acute Myeloid Leukemia. JCI Insight 2016, 1, e85630. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Krämer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus Salvage Chemotherapy in Relapsed or Refractory FLT3-ITD Acute Myeloid Leukaemia (QuANTUM-R): A Multicentre, Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 984–997. [Google Scholar] [CrossRef]
- Cortes, J.E.; Tallman, M.S.; Schiller, G.J.; Trone, D.; Gammon, G.; Goldberg, S.L.; Perl, A.E.; Marie, J.-P.; Martinelli, G.; Kantarjian, H.M.; et al. Phase 2b Study of 2 Dosing Regimens of Quizartinib Monotherapy in FLT3-ITD–Mutated, Relapsed or Refractory AML. Blood 2018, 132, 598–607. [Google Scholar] [CrossRef]
- Home—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ (accessed on 15 May 2022).
- Xuan, L.; Wang, Y.; Huang, F.; Fan, Z.; Xu, Y.; Sun, J.; Xu, N.; Deng, L.; Li, X.; Liang, X.; et al. Sorafenib Maintenance in Patients with FLT3-ITD Acute Myeloid Leukaemia Undergoing Allogeneic Haematopoietic Stem-Cell Transplantation: An Open-Label, Multicentre, Randomised Phase 3 Trial. Lancet Oncol. 2020, 21, 1201–1212. [Google Scholar] [CrossRef]
- Chevallier, P. Phase 2 Study of Ponatinib (Iclusig) for Prevention of Relapse after Allogeneic Stem Cell Transplantation (Allo-SCT) in FLT3-ITD AML Patients: The PONALLO Trial; Clinicaltrials.Gov: Bethesda, MA, USA, 2021.
- M.D. Anderson Cancer Center. A Phase II Study of the Combination of Decitabine, Venetoclax, and Ponatinib in Patients with Philadelphia Chromosome-Positive Acute Myeloid Leukemia or Myeloid Blast Phase Chronic Myelogenous Leukemia; Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- Incyte Biosciences International Sàrl. An Open-Label, Single-Arm, Phase 1/2 Study Evaluating the Safety and Efficacy of Ponatinib for the Treatment of Recurrent or Refractory Leukemias or Solid Tumors in Pediatric Participants; Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- Wang, E. Phase 3, Multicenter, Open-Label Study of Gilteritinib, Gilteritinib Plus Azacitidine, or Azacitidine Alone in Newly Diagnosed FLT3 Mutated (FLT3mut+) Acute Myeloid Leukemia (AML) Patients Ineligible for Intensive Induction Chemotherapy; ASH: Washington, DC, USA, 2020. [Google Scholar]
- Astellas Pharma Global Development, Inc. A Phase 3 Multicenter, Open-Label, Randomized Study of ASP2215 (Gilteritinib), Combination of ASP2215 Plus Azacitidine and Azacitidine Alone in the Treatment of Newly Diagnosed Acute Myeloid Leukemia with FLT3 Mutation in Patients Not Eligible for Intensive Induction Chemotherapy; Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- Astellas Pharma Global Development, Inc. A Multi-Center, Randomized, Double-Blind, Placebo-Controlled Phase III Trial of the FLT3 Inhibitor Gilteritinib Administered as Maintenance Therapy Following Allogeneic Transplant for Patients with FLT3/ITD AML; Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- Astellas Pharma Global Development, Inc. A Phase 2 Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial of the FLT3 Inhibitor Gilteritinib (ASP2215) Administered as Maintenance Therapy Following Induction/Consolidation Therapy for Subjects with FLT3/ITD AML in First Complete Remission; Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- PrECOG, LLC. Randomized Trial of Gilteritinib vs. Midostaurin in FLT3 Mutated Acute Myeloid Leukemia (AML); Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- Oran, B.; Ciurea, S.O.; Marin, D.; McCarty, J.M.; Bashir, Q.; Ahmed, S.; Olson, A.L.; Popat, U.; Nieto, Y.; Kebriaei, P.; et al. Safety Analysis of Intra-Patient Dose- Study of Crenolanib Maintenance Therapy in Patients with FLT3 Mutant AML Following Allogeneic Hematopoietic Stem Cell Transplant. Blood 2018, 132, 3426. [Google Scholar] [CrossRef]
- Arog Pharmaceuticals, Inc. Phase III Randomized Study of Crenolanib Versus Midostaurin Administered Following Induction Chemotherapy and Consolidation Therapy in Newly Diagnosed Subjects with FLT3 Mutated Acute Myeloid Leukemia; Clinicaltrials.Gov: Bethesda, MA, USA, 2020.
- Arog Pharmaceuticals, Inc. Phase III Randomized, Double-Blind, Placebo-Controlled Study Investigating the Efficacy of the Addition of Crenolanib to Salvage Chemotherapy Versus Salvage Chemotherapy Alone in Subjects ≤75 Years of Age with Relapsed/Refractory FLT3 Mutated Acute Myeloid Leukemia; Clinicaltrials.Gov: Bethesda, MA, USA, 2021.
- Meryx, Inc. An Open Label Evaluation Phase 1 Trial of the Safety and Pharmacokinetics of MRX-2843 in Adolescents and Adults with Relapsed/Refractory Acute Myeloid Leukemia, Acute Lymphoblastic Leukemia, or Mixed Phenotype Acute Leukemia; Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- Betta Pharmaceuticals Co., Ltd. Phase I/II Clinical Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Effectiveness of MRX2843 Tablets in Patients with Relapsed/Refractory Acute Myeloid Leukemia; Clinicaltrials.Gov: Bethesda, MA, USA, 2021.
- ASH. FDA Declines to Approve Quizartinib for FLT3-Mutated Acute Myeloid Leukemia; ASH: Washington, DC, USA, 2021. [Google Scholar]
- Daiichi Sankyo, Inc. A Phase 3, Double-Blind, Placebo-Controlled Study of Quizartinib Administered in Combination with Induction and Consolidation Chemotherapy, and Administered as Continuation Therapy in Subjects 18 to 75 Years Old with Newly Diagnosed FLT3-ITD (+) Acute Myeloid Leukemia (QuANTUM First); Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- Lonetti, A.; Pession, A.; Masetti, R. Targeted Therapies for Pediatric AML: Gaps and Perspective. Front. Pediatr. 2019, 7, 463. [Google Scholar] [CrossRef]
- Knight, T.E.; Ge, Y.; Taub, J.W.; Hitzler, J.; Krueger, J. When It Comes to Drug Access, Should Children Be Considered Small Adults? Countering Coverage Denials of FLT3 Inhibitors in Children with FLT3-ITD AML. Pediatr. Blood Cancer 2021, 68, e29278. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Brown, P.A.; Gerbing, R.B.; Fox, E.; Choi, J.K.; Fisher, B.T.; Hirsch, B.A.; Kahwash, S.; Levine, J.E.; et al. Sorafenib in Combination with Standard Chemotherapy for Children with High Allelic Ratio FLT3/ITD+ AML Improves Event-Free Survival and Reduces Relapse Risk: A Report from the Children’s Oncology Group Protocol AAML1031. Blood 2019, 134, 292. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Gerbing, R.; Brown, P.; Fox, E.; Choi, J.; Fisher, B.; Hirsch, B.; Kahwash, S.; Getz, K.; et al. Sorafenib in Combination with Standard Chemotherapy for Children with High Allelic Ratio FLT3/ITD+ Acute Myeloid Leukemia: A Report from the Children’s Oncology Group Protocol AAML1031. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 18. [Google Scholar] [CrossRef] [PubMed]
- Zwaan, C.M.; Söderhäll, S.; Brethon, B.; Luciani, M.; Rizzari, C.; Stam, R.W.; Besse, E.; Dutreix, C.; Fagioli, F.; Ho, P.A.; et al. A Phase 1/2, Open-Label, Dose-Escalation Study of Midostaurin in Children with Relapsed or Refractory Acute Leukaemia. Br. J. Haematol. 2019, 185, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.M.; Cassar, J.; Eckroth, E.; Malvar, J.; Sposto, R.; Gaynon, P.; Chang, B.H.; Gore, L.; August, K.; Pollard, J.A.; et al. A Phase I Study of Quizartinib Combined with Chemotherapy in Relapsed Childhood Leukemia: A Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4014–4022. [Google Scholar] [CrossRef] [Green Version]
- Novartis Pharmaceuticals. A Phase II, Open-Label, Single Arm Study to Evaluate the Safety, Efficacy, and Pharmacokinetics of Twice Daily Midostaurin (PKC412) Combined with Standard Chemotherapy and as a Single Agent Post-Consolidation Therapy in Children with Untreated FLT3-Mutated AML; Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- Daiichi Sankyo, Inc. A Phase 1/2, Multicenter, Dose-Escalating Study to Evaluate the Safety, Pharmacokinetics, Pharmacodynamics, and Efficacy Of Quizartinib Administered in Combination with Re-Induction Chemotherapy, and as a Single-Agent Continuation Therapy, in Pediatric Relapsed/Refractory AML Subjects Aged 1 Month to <18 Years (and Young Adults Aged up to 21 Years) with FLT3-ITD Mutations; Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- Inaba, H.; van Oosterwijk, J.G.; Panetta, J.C.; Li, L.; Buelow, D.R.; Blachly, J.S.; Shurtleff, S.; Pui, C.-H.; Ribeiro, R.C.; Rubnitz, J.E.; et al. Preclinical and Pilot Study of Type I FLT3 Tyrosine Kinase Inhibitor, Crenolanib, with Sorafenib in Acute Myeloid Leukemia and FLT3-Internal Tandem Duplication. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 2536–2546. [Google Scholar] [CrossRef] [PubMed]
- Astellas Pharma Global Development, Inc. A Phase 1/2, Multicenter, Open-Label, Single Arm, Dose Escalation and Expansion Study of Gilteritinib (ASP2215) Combined with Chemotherapy in Children, Adolescents and Young Adults with FMS-like Tyrosine Kinase 3 (FLT3)/Internal Tandem Duplication (ITD) Positive Relapsed or Refractory Acute Myeloid Leukemia (AML); Clinicaltrials.Gov: Bethesda, MA, USA, 2022.
- Pollard, J.A.; Alonzo, T.A.; Loken, M.; Gerbing, R.B.; Ho, P.A.; Bernstein, I.D.; Raimondi, S.C.; Hirsch, B.; Franklin, J.; Walter, R.B.; et al. Correlation of CD33 Expression Level with Disease Characteristics and Response to Gemtuzumab Ozogamicin Containing Chemotherapy in Childhood AML. Blood 2012, 119, 3705–3711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, J.A.; Loken, M.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Aplenc, R.; Bernstein, I.D.; Gamis, A.S.; Alonzo, T.A.; Meshinchi, S. CD33 Expression and Its Association with Gemtuzumab Ozogamicin Response: Results from the Randomized Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2016, 34, 747–755. [Google Scholar] [CrossRef]
- Tarlock, K.; Alonzo, T.A.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Sung, L.; Pollard, J.A.; Aplenc, R.; Loken, M.R.; Gamis, A.S.; et al. Gemtuzumab Ozogamicin Reduces Relapse Risk in FLT3/ITD Acute Myeloid Leukemia: A Report from the Children’s Oncology Group. Clin. Cancer Res. 2016, 22, 1951–1957. [Google Scholar] [CrossRef] [Green Version]
- Röllig, C. Gemtuzumab Ozogamicin Plus Midostaurin in Combination with Standard Intensive Induction Therapy in Newly Diagnosed AML: Results from a Phase-I Study; ASH: Washington, DC, USA, 2021. [Google Scholar]
- Chen, F.; Ishikawa, Y.; Akashi, A.; Naoe, T.; Kiyoi, H. Co-Expression of Wild-Type FLT3 Attenuates the Inhibitory Effect of FLT3 Inhibitor on FLT3 Mutated Leukemia Cells. Oncotarget 2016, 7, 47018–47032. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Sexauer, A.; Levis, M. Bone Marrow Stroma-Mediated Resistance to FLT3 Inhibitors in FLT3-ITD AML Is Mediated by Persistent Activation of Extracellular Regulated Kinase. Br. J. Haematol. 2014, 164, 61–72. [Google Scholar] [CrossRef]
- Sato, T.; Yang, X.; Knapper, S.; White, P.; Smith, B.D.; Galkin, S.; Small, D.; Burnett, A.; Levis, M. FLT3 Ligand Impedes the Efficacy of FLT3 Inhibitors In Vitro and In Vivo. Blood 2011, 117, 3286–3293. [Google Scholar] [CrossRef] [Green Version]
- Cerchione, C.; Peleteiro Raíndo, A.; Mosquera Orgueira, A.; Mosquera Torre, A.; Bao Pérez, L.; Marconi, G.; Isidori, A.; Pérez Encinas, M.M.; Martinelli, G. Safety of FLT3 Inhibitors in Patients with Acute Myeloid Leukemia. Expert Rev. Hematol. 2021, 14, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kankam, M.; Trone, D.; Gammon, G. Effects of CYP3A Inhibitors on the Pharmacokinetics of Quizartinib, a Potent and Selective FLT3 Inhibitor, and Its Active Metabolite. Br. J. Clin. Pharmacol. 2019, 85, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Dutreix, C.; Munarini, F.; Lorenzo, S.; Roesel, J.; Wang, Y. Investigation into CYP3A4-Mediated Drug-Drug Interactions on Midostaurin in Healthy Volunteers. Cancer Chemother. Pharmacol. 2013, 72, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-T.; Hernandez, D.; Alonso, S.; Gao, M.; Su, M.; Ghiaur, G.; Levis, M.J.; Jones, R.J. Role of CYP3A4 in Bone Marrow Microenvironment–Mediated Protection of FLT3/ITD AML from Tyrosine Kinase Inhibitors. Blood Adv. 2019, 3, 908–916. [Google Scholar] [CrossRef]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.-T.; Levis, M.; Small, D. Prolonged Exposure to FLT3 Inhibitors Leads to Resistance via Activation of Parallel Signaling Pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidel, F.; Solem, F.K.; Breitenbuecher, F.; Lipka, D.B.; Kasper, S.; Thiede, M.H.; Brandts, C.; Serve, H.; Roesel, J.; Giles, F.; et al. Clinical Resistance to the Kinase Inhibitor PKC412 in Acute Myeloid Leukemia by Mutation of Asn-676 in the FLT3 Tyrosine Kinase Domain. Blood 2006, 107, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.D.; Zimmerman, E.I.; Wang, Y.-D.; Orwick, S.; Zatechka, D.S.; Buaboonnam, J.; Neale, G.A.; Olsen, S.R.; Enemark, E.J.; Shurtleff, S.; et al. Emergence of Polyclonal FLT3 Tyrosine Kinase Domain Mutations during Sequential Therapy with Sorafenib and Sunitinib in FLT3-ITD-Positive Acute Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 5758–5768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, X.; Ma, J.; Knight, T.; Su, Y.; Edwards, H.; Polin, L.; Li, J.; Kushner, J.; Dzinic, S.H.; White, K.; et al. The Combination of CUDC-907 and Gilteritinib Shows Promising In Vitro and In Vivo Antileukemic Activity against FLT3-ITD AML. Blood Cancer J. 2021, 11, 111. [Google Scholar] [CrossRef]
- Li, X.; Su, Y.; Madlambayan, G.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Ma, J.; Knight, T.; et al. Antileukemic Activity and Mechanism of Action of the Novel PI3K and Histone Deacetylase Dual Inhibitor CUDC-907 in Acute Myeloid Leukemia. Haematologica 2019, 104, 2225–2240. [Google Scholar] [CrossRef] [Green Version]
- Park, I.-K.; Mundy-Bosse, B.; Whitman, S.P.; Zhang, X.; Warner, S.L.; Bearss, D.J.; Blum, W.; Marcucci, G.; Caligiuri, M.A. Receptor Tyrosine Kinase Axl Is Required for Resistance of Leukemic Cells to FLT3-Targeted Therapy in Acute Myeloid Leukemia. Leukemia 2015, 29, 2382–2389. [Google Scholar] [CrossRef]
- Knight, T.; Luedtke, D.; Edwards, H.; Taub, J.W.; Ge, Y. A Delicate Balance—The BCL-2 Family and Its Role in Apoptosis, Oncogenesis, and Cancer Therapeutics. Biochem. Pharmacol. 2019, 162, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Knight, T.; Edwards, H.; Taub, J.W.; Ge, Y. Evaluating Venetoclax and Its Potential in Treatment-Naïve Acute Myeloid Leukemia. Cancer Manag. Res. 2019, 11, 3197–3213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Zhao, S.; Qiao, X.; Knight, T.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Wang, G.; et al. Inhibition of Bcl-2 Synergistically Enhances the Antileukemic Activity of Midostaurin and Gilteritinib in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6815–6826. [Google Scholar] [CrossRef]
- Liu, F.; Knight, T.; Su, Y.; Edwards, H.; Wang, G.; Wang, Y.; Taub, J.W.; Lin, H.; Sun, L.; Ge, Y. Venetoclax Synergistically Enhances the Anti-Leukemic Activity of Vosaroxin against Acute Myeloid Leukemia Cells Ex Vivo. Target. Oncol. 2019, 14, 351–364. [Google Scholar] [CrossRef]
- Li, X.; Su, Y.; Hege, K.; Madlambayan, G.; Edwards, H.; Knight, T.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; et al. The HDAC and PI3K Dual Inhibitor CUDC-907 Synergistically Enhances the Antileukemic Activity of Venetoclax in Preclinical Models of Acute Myeloid Leukemia. Haematologica 2021, 106, 1262–1277. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wu, S.; Liu, S.; Li, X.; Gai, Y.; Lin, H.; Wang, Y.; Edwards, H.; Ge, Y.; Wang, G. Venetoclax Enhances DNA Damage Induced by XPO1 Inhibitors: A Novel Mechanism Underlying the Synergistic Antileukaemic Effect in Acute Myeloid Leukaemia. J. Cell. Mol. Med. 2022, 26, 2646–2657. [Google Scholar] [CrossRef]
- Niu, X.; Rothe, K.; Chen, M.; Grasedieck, S.; Li, R.; Nam, S.-E.; Zhang, X.; Novakovskiy, G.E.; Ahn, Y.-H.; Maksakova, I.; et al. Targeting AXL Kinase Sensitizes Leukemic Stem and Progenitor Cells to Venetoclax Treatment in Acute Myeloid Leukemia. Blood 2021, 137, 3641–3655. [Google Scholar] [CrossRef]
- Short, N.J.; Borthakur, G.; Pemmaraju, N.; Dinardo, C.D.; Kadia, T.M.; Jabbour, E.; Konopleva, M.; Macaron, W.; Ning, J.; Ma, J.; et al. A Multi-Arm Phase Ib/II Study Designed for Rapid, Parallel Evaluation of Novel Immunotherapy Combinations in Relapsed/Refractory Acute Myeloid Leukemia. Leuk. Lymphoma 2022, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Konopleva, M.Y. How We Incorporate Venetoclax in Treatment Regimens for Acute Myeloid Leukemia. Cancer J. Sudbury Mass 2022, 28, 2–13. [Google Scholar] [CrossRef]
- Garcia, J.S.; Kim, H.T.; Murdock, H.M.; Cutler, C.S.; Brock, J.; Gooptu, M.; Ho, V.T.; Koreth, J.; Nikiforow, S.; Romee, R.; et al. Adding Venetoclax to Fludarabine/Busulfan RIC Transplant for High-Risk MDS and AML Is Feasible, Safe, and Active. Blood Adv. 2021, 5, 5536–5545. [Google Scholar] [CrossRef] [PubMed]
- M.D. Anderson Cancer Center. A Phase Ib/II Study of Venetoclax in Combination with Quizartinib in FLT3-Mutated Acute Myelogenous Leukemia (AML); Clinicaltrials.Gov: Bethesda, MA, USA, 2020.
- M.D. Anderson Cancer Center. A Phase I/II Study of Azacitidine, Venetoclax, and Gilteritinib for Patients with Acute Myeloid Leukemia or High-Risk Myelodysplastic Syndrome with an Activating FLT3 Mutation; Clinicaltrials.Gov: Bethesda, MA, USA, 2021.
- M.D. Anderson Cancer Center. A Phase I/II Study of ASTX727, Venetoclax, and Gilteritinib for Patients with Acute Myeloid Leukemia or High-Risk Myelodysplastic Syndrome with an Activating FLT3 Mutation; Clinicaltrials.Gov: Bethesda, MA, USA, 2022.


{kind=link}
{kind=link}
| Type 1 FLT3 Inhibitors (Inhibition of Both Active and Inactive FLT3 Confirmation) | Type 2 FLT3 Inhibitors (Inhibition of Inactive Conformation Only) | |
|---|---|---|
| First Generation FLT3 Inhibitors | * Midostaurin [80,81,82,83] Lestaurtinib [84,85] Sunitinib [86] | Sorafenib [87,88,89,90] Pexidartinib [91] Ponatinib [92,93,94,95] |
| Second Generation FLT3 Inhibitors | ** Gilteritinib [96,97,98,99,100,101] Crenolanib [67,102,103,104,105] MRX-2843 [106,107,108] | Quizartinib [109,110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knight, T.E.; Edwards, H.; Meshinchi, S.; Taub, J.W.; Ge, Y. “FLipping” the Story: FLT3-Mutated Acute Myeloid Leukemia and the Evolving Role of FLT3 Inhibitors. Cancers 2022, 14, 3398. https://doi.org/10.3390/cancers14143398
Knight TE, Edwards H, Meshinchi S, Taub JW, Ge Y. “FLipping” the Story: FLT3-Mutated Acute Myeloid Leukemia and the Evolving Role of FLT3 Inhibitors. Cancers. 2022; 14(14):3398. https://doi.org/10.3390/cancers14143398
Chicago/Turabian StyleKnight, Tristan E., Holly Edwards, Soheil Meshinchi, Jeffrey W. Taub, and Yubin Ge. 2022. "“FLipping” the Story: FLT3-Mutated Acute Myeloid Leukemia and the Evolving Role of FLT3 Inhibitors" Cancers 14, no. 14: 3398. https://doi.org/10.3390/cancers14143398