
The Role of PIM Kinases in Pediatric Solid Tumors


Abstract
Simple Summary
Abstract
1. Introduction
2. PIM Kinase Family
3. PIM Kinase Structure
4. PIM Kinases Expression and Regulation
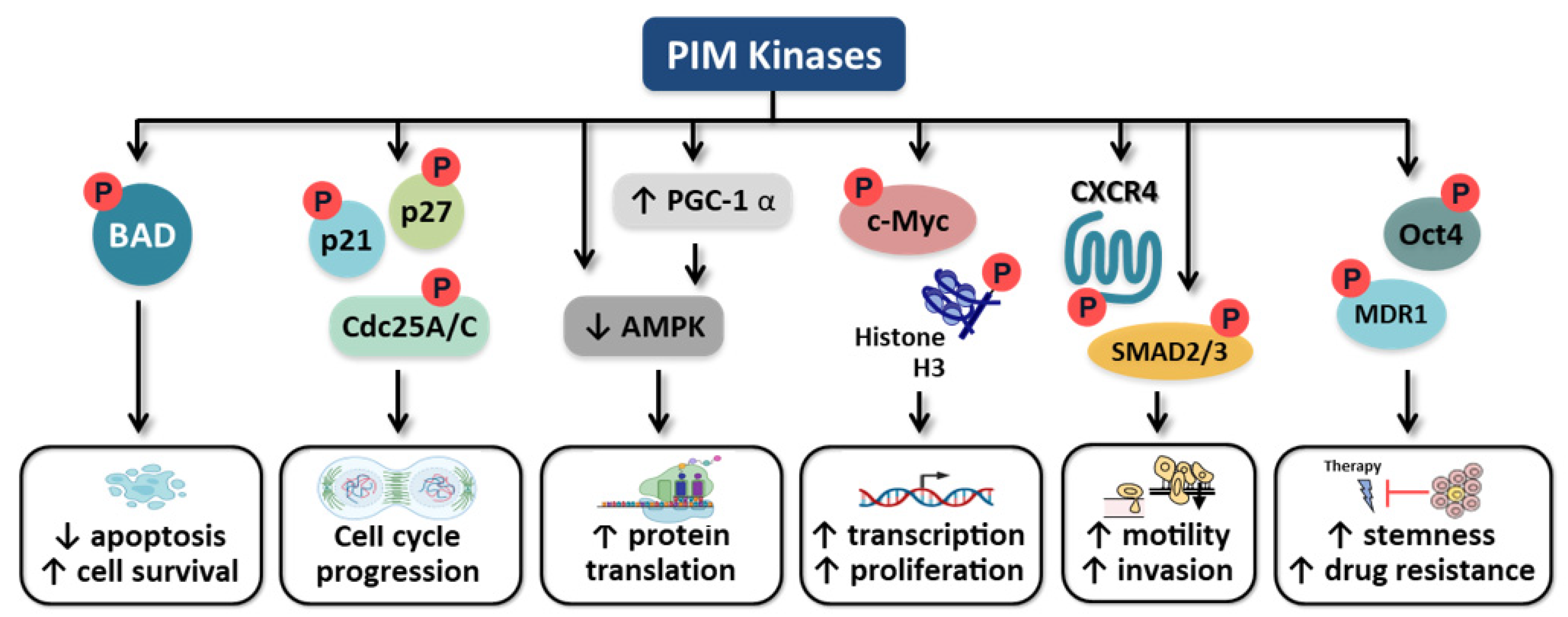
5. Role of PIM Kinases in Carcinogenesis
5.1. Apoptosis
5.2. Cell Cycle
5.3. C-Myc Regulation
5.4. Motility
5.5. Cancer Cell Stemness
6. Role of PIM Kinases in Pediatric Malignancies
6.1. Hepatoblastoma
6.2. Neuroblastoma
6.3. Osteosarcoma
7. Pharmacologic Inhibition of PIM Kinases
7.1. Therapeutic Agents
7.2. Clinical Trials
7.3. Combination Therapies including PIM Kinase Inhibitors
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aziz, A.u.R.; Farid, S.; Qin, K.; Wang, H.; Liu, B. Regulation of insulin resistance and glucose metabolism by interaction of PIM kinases and insulin receptor substrates. Arch. Physiol. Biochem. 2020, 126, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Carlson, D.A.; Singer, M.R.; Sutherland, C.; Redondo, C.; Alexander, L.T.; Hughes, P.F.; Knapp, S.; Gurley, S.B.; Sparks, M.A.; MacDonald, J.A.; et al. Targeting Pim Kinases and DAPK3 to Control Hypertension. Cell Chem. Biol. 2018, 25, 1195–1207.e1132. [Google Scholar] [CrossRef] [PubMed]
- Maney, N.J.; Lemos, H.; Barron-Millar, B.; Carey, C.; Herron, I.; Anderson, A.E.; Mellor, A.L.; Isaacs, J.D.; Pratt, A.G. Pim Kinases as Therapeutic Targets in Early Rheumatoid Arthritis. Arthritis Rheumatol. 2021, 73, 1820–1830. [Google Scholar] [CrossRef] [PubMed]
- Nawijn, M.C.; Alendar, A.; Berns, A. For better or for worse: The role of Pim oncogenes in tumorigenesis. Nat. Rev. Cancer 2011, 11, 23–34. [Google Scholar] [CrossRef]
- Zhu, N.; Ramirez, L.M.; Lee, R.L.; Magnuson, N.S.; Bishop, G.A.; Gold, M.R. CD40 signaling in B cells regulates the expression of the Pim-1 kinase via the NF-kappa B pathway. J. Immunol. 2002, 168, 744–754. [Google Scholar] [CrossRef]
- Fox, C.J.; Hammerman, P.S.; Cinalli, R.M.; Master, S.R.; Chodosh, L.A.; Thompson, C.B. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 2003, 17, 1841–1854. [Google Scholar] [CrossRef]
- Fujii, C.; Nakamoto, Y.; Lu, P.; Tsuneyama, K.; Popivanova, B.K.; Kaneko, S.; Mukaida, N. Aberrant expression of serine/threonine kinase Pim-3 in hepatocellular carcinoma development and its role in the proliferation of human hepatoma cell lines. Int. J. Cancer 2005, 114, 209–218. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, K.; Dai, B.; Guo, Z.; Jiang, T.; Chen, H.; Qiu, Y. The 44 kDa Pim-1 kinase directly interacts with tyrosine kinase Etk BMX and protects human prostate cancer cells from apoptosis induced by chemotherapeutic drugs. Oncogene 2006, 25, 70–78. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Mukaida, N. Pathophysiological roles of Pim-3 kinase in pancreatic cancer development and progression. World J. Gastroenterol. WJG 2014, 20, 9392–9404. [Google Scholar] [CrossRef]
- Qian, K.C.; Wang, L.; Hickey, E.R.; Studts, J.; Barringer, K.; Peng, C.; Kronkaitis, A.; Li, J.; White, A.; Mische, S.; et al. Structural Basis of Constitutive Activity and a Unique Nucleotide Binding Mode of Human Pim-1 Kinase. J. Biol. Chem. 2005, 280, 6130–6137. [Google Scholar] [CrossRef]
- Adam, K.; Lambert, M.; Lestang, E.; Champenois, G.; Dusanter-Fourt, I.; Tamburini, J.; Bouscary, D.; Lacombe, C.; Zermati, Y.; Mayeux, P. Control of Pim2 kinase stability and expression in transformed human haematopoietic cells. Biosci. Rep. 2015, 35, e00274. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Russo, S.; Amos, A.; Pagano, N.; Bregman, H.; Debreczeni, J.E.; Lee, W.H.; von Delft, F.; Meggers, E.; Knapp, S. Crystal structure of the PIM2 kinase in complex with an organoruthenium inhibitor. PLoS ONE 2009, 4, e7112. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, T.; Wang, T.; You, L.; Zhao, Y. PIM kinases: An overview in tumors and recent advances in pancreatic cancer. Future Oncol. 2014, 10, 865–876. [Google Scholar] [CrossRef]
- Warfel, N.A.; Kraft, A.S. PIM kinase (and Akt) biology and signaling in tumors. Pharmacol. Ther. 2015, 151, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Domen, J.; Van Der Lugt, N.M.T.; Laird, P.W.; Saris, C.J.M.; Berns, A. Analysis of Pim-1 function in mutant mice. Leukemia 1993, 7, S108–S112. [Google Scholar] [PubMed]
- Amson, R.; Sigaux, F.; Przedborski, S.; Flandrin, G.; Givol, D.; Telerman, A. The Human Protooncogene Product P33pim is Expressed during Fetal Hematopoiesis and in Diverse Leukemias. Proc. Natl. Acad. Sci. USA 1989, 86, 8857–8861. [Google Scholar] [CrossRef]
- Domen, J.; Van Der Lugt, N.M.T.; Acton, D.; Laird, P.W.; Berns, A.; Linders, K. Pim-1 levels determine the size of early B lymphoid compartments in bone marrow. J. Exp. Med. 1993, 178, 1665–1673. [Google Scholar] [CrossRef]
- Domen, J.; Van Der Lugt, N.M.T.; Laird, P.W.; Saris, C.J.M.; Clarke, A.R.; Hooper, M.L.; Berns, A. Impaired Interleukin-3 Response in Pim-1-Deficient Bone Marrow-Derived Mast Cells. Blood 1993, 82, 1445–1452. [Google Scholar] [CrossRef]
- Blanco-Aparicio, C.; Carnero, A. Pim kinases in cancer: Diagnostic, prognostic and treatment opportunities. Biochem. Pharmacol. 2013, 85, 629–643. [Google Scholar] [CrossRef]
- Mikkers, H.; Nawijn, M.; Allen, J.; Brouwers, C.; Verhoeven, E.; Jonkers, J.; Berns, A. Mice Deficient for All PIM Kinases Display Reduced Body Size and Impaired Responses to Hematopoietic Growth Factors. Mol. Cell. Biol. 2004, 24, 6104–6115. [Google Scholar] [CrossRef]
- Allen, J.D.; Verhoeven, E.; Domen, J.; Van Der Valk, M.; Berns, A. Pim-2 transgene induces lymphoid tumors, exhibiting potent synergy with c-myc. Oncogene 1997, 15, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Dautry, F.; Weil, D.; Yu, J.; Dautry-Varsat, A. Regulation of pim and myb mRNA accumulation by interleukin 2 and interleukin 3 in murine hematopoietic cell lines. J. Biol. Chem. 1988, 263, 17615–17620. [Google Scholar] [CrossRef]
- Stout, B.A.; Bates, M.E.; Liu, L.Y.; Farrington, N.N.; Bertics, P.J. IL-5 and Granulocyte-Macrophage Colony-Stimulating Factor Activate STAT3 and STAT5 and Promote Pim-1 and Cyclin D3 Protein Expression in Human Eosinophils. J. Immunol. 2004, 173, 6409–6417. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Wang, Y.; Wu, C.L.; Liu, K.Y.; Chen, H.; Mao, Z.B. PIM-1 modulates cellular senescence and links IL-6 signaling to heterochromatin formation. Aging Cell 2014, 13, 879–889. [Google Scholar] [CrossRef]
- Aho, T.L.T.; Lund, R.J.; Ylikoski, E.K.; Matikainen, S.; Lahesmaa, R.; Koskinen, P.J. Expression of human pim family genes is selectively up-regulated by cytokines promoting T helper type 1, but not T helper type 2, cell differentiation. Immunology 2005, 116, 82–88. [Google Scholar] [CrossRef]
- Miura, O.; Miura, Y.; Nakamura, N.; Quelle, F.W.; Witthuhn, B.A.; Ihle, J.N.; Aoki, N. Induction of tyrosine phosphorylation of Vav and expression of Pim-1 correlates with Jak2-mediated growth signaling from the erythropoietin receptor. Blood 1994, 84, 4135–4141. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Horie, M.; Broxmeyer, H.E. Transcriptional Induction of pim-1 Protein Kinase Gene Expression by Interferon γ and Posttranscriptional Effects on Costimulation with Steel Factor. Blood 1995, 85, 3494–3502. [Google Scholar] [CrossRef]
- Krishnan, N.; Pan, H.; Buckley, D.J.; Buckley, A. Prolactin-regulated pim-1 transcription: Identification of critical promoter elements and Akt signaling. Endocrine 2003, 20, 123–130. [Google Scholar] [CrossRef]
- Lilly, M.; Tiep, L.E.; Holland, P.; Hendrickson, S.L. Sustained expression of the pim-1 kinase is specifically induced in myeloid cells by cytokines whose receptors are structurally related. Oncogene 1992, 7, 727–732. [Google Scholar]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef]
- Peltola, K.J.; Paukku, K.; Aho, T.L.T.; Ruuska, M.; Silvennoinen, O.; Koskinen, P.J. Pim-1 kinase inhibits STAT5-dependent transcription via its interactions with SOCS1 and SOCS3. Blood 2004, 103, 3744–3750. [Google Scholar] [CrossRef] [PubMed]
- Uddin, N.; Kim, R.K.; Yoo, K.C.; Kim, Y.H.; Cui, Y.H.; Kim, I.G.; Suh, Y.; Lee, S.J. Persistent activation of STAT3 by PIM2-driven positive feedback loop for epithelial-mesenchymal transition in breast cancer. Cancer Sci. 2015, 106, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Peet, G.W.; Balzarano, D.; Li, X.; Massa, P.; Barton, R.W.; Marcu, K.B. Novel NEMO/IkappaB kinase and NF-kappa B target genes at the pre-B to immature B cell transition. J. Biol. Chem. 2001, 276, 18579–18590. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hamza, M.S.; Leong, H.S.; Lim, C.B.; Pan, Y.F.; Cheung, E.; Soo, K.C.; Iyer, N.G. Kruppel-like factor 5 modulates p53-independent apoptosis through Pim1 survival kinase in cancer cells. Oncogene 2008, 27, 1–8. [Google Scholar] [CrossRef][Green Version]
- Hu, Y.-L.; Passegué, E.; Fong, S.; Largman, C.; Lawrence, H.J. Evidence that the Pim1 kinase gene is a direct target of HOXA9. Blood 2007, 109, 4732–4738. [Google Scholar] [CrossRef]
- Yu, Z.; Zhao, X.; Ge, Y.; Zhang, T.; Huang, L.; Zhou, X.; Xie, L.; Liu, J.; Huang, G. A regulatory feedback loop between HIF-1α and PIM2 in HepG2 cells. PLoS ONE 2014, 9, e88301. [Google Scholar] [CrossRef]
- Chen, J.; Kobayashi, M.; Darmanin, S.; Qiao, Y.; Gully, C.; Zhao, R.; Kondo, S.; Wang, H.; Wang, H.; Yeung, S.-C.J.; et al. Hypoxia-Mediated Up-Regulation of Pim-1 Contributes to Solid Tumor Formation. Am. J. Pathol. 2009, 175, 400–411. [Google Scholar] [CrossRef]
- Hoover, D.S.; Wingett, D.G.; Zhang, J.; Reeves, R.; Magnuson, N.S. Pim-1 protein expression is regulated by its 5’-untranslated region and translation initiation factor eIF-4E. Cell Growth Differ. 1997, 8, 1371–1380. [Google Scholar]
- Culjkovic, B.; Topisirovic, I.; Skrabanek, L.; Ruiz-Gutierrez, M.; Borden, K.L.B. eIF4E Is a Central Node of an RNA Regulon That Governs Cellular Proliferation. J. Cell Biol. 2006, 175, 415–426. [Google Scholar] [CrossRef]
- Du, J.; Zheng, X.I.; Cai, S.; Zhu, Z.; Tan, J.; Hu, B.I.N.; Huang, Z.; Jiao, H. MicroRNA-506 participates in pancreatic cancer pathogenesis by targeting PIM3. Mol. Med. Rep. 2015, 12, 5121–5126. [Google Scholar] [CrossRef]
- Liang, C.; Yu, X.-J.; Guo, X.-Z.; Sun, M.-H.; Wang, Z.; Song, Y.; Ni, Q.-X.; Li, H.-Y.; Mukaida, N.; Li, Y.-Y. MicroRNA-33a-mediated downregulation of Pim-3 kinase expression renders human pancreatic cancer cells sensitivity to gemcitabine. Oncotarget 2015, 6, 14440–14455. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, I.; Zbinden, M.; Byles, V.; Torrence, M.; Manning, B.D. mTORC1 suppresses PIM3 expression via miR-33 encoded by the SREBP loci. Sci. Rep. 2017, 7, 16112. [Google Scholar] [CrossRef]
- Saris, C.J.; Domen, J.; Berns, A. The pim-1 oncogene encodes two related protein-serine/threonine kinases by alternative initiation at AUG and CUG. EMBO J. 1991, 10, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Hittelman, W.; Nagarajan, L. Ubiquitous Expression and Cell Cycle Regulation of the Protein Kinase PIM-1. Arch. Biochem. Biophys. 1996, 330, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Chen, X.P.; Vuong, B.Q.; Fay, S.; Rothman, P.B. Protein Phosphatase 2A Regulates the Stability of Pim Protein Kinases. J. Biol. Chem. 2003, 278, 4800–4805. [Google Scholar] [CrossRef]
- Ma, J.; Arnold, H.K.; Lilly, M.B.; Sears, R.C.; Kraft, A.S. Negative regulation of Pim-1 protein kinase levels by the B56beta subunit of PP2A. Oncogene 2007, 26, 5145–5153. [Google Scholar] [CrossRef]
- Wang, Y.; Xiu, J.; Ren, C.; Yu, Z. Protein kinase PIM2: A simple PIM family kinase with complex functions in cancer metabolism and therapeutics. J. Cancer 2021, 12, 2570–2581. [Google Scholar] [CrossRef]
- Shah, N.; Pang, B.; Yeoh, K.-G.; Thorn, S.; Chen, C.S.; Lilly, M.B.; Salto-Tellez, M. Potential roles for the PIM1 kinase in human cancer—A molecular and therapeutic appraisal. Eur. J. Cancer 2008, 44, 2144–2151. [Google Scholar] [CrossRef]
- Bachmann, M.; Möröy, T. The serine/threonine kinase Pim-1. Int. J. Biochem. Cell Biol. 2005, 37, 726–730. [Google Scholar] [CrossRef]
- Guo, S.; Mao, X.; Chen, J.; Huang, B.; Jin, C.; Xu, Z.; Qiu, S. Overexpression of Pim-1 in bladder cancer. J. Exp. Clin. Cancer Res. 2010, 29, 161. [Google Scholar] [CrossRef]
- Nga, M.E.; Swe, N.N.M.; Chen, K.T.; Shen, L.; Lilly, M.B.; Chan, S.P.; Salto-Tellez, M.; Das, K. PIM-1 kinase expression in adipocytic neoplasms: Diagnostic and biological implications. Int. J. Exp. Pathol. 2010, 91, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Möröy, T.; Grzeschiczek, A.; Petzold, S.; Hartmann, K.U. Expression of a Pim-1 Transgene Accelerates Lymphoproliferation and Inhibits Apoptosis in lpr/lpr Mice. Proc. Natl. Acad. Sci. USA 1993, 90, 10734–10738. [Google Scholar] [CrossRef] [PubMed]
- Rathi, A.; Kumar, D.; Hasan, G.M.; Haque, M.M.; Hassan, M.I. Therapeutic targeting of PIM KINASE signaling in cancer therapy: Structural and clinical prospects. Biochim. Et Biophys. Acta. Gen. Subj. 2021, 1865, 129995. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Popivanova, B.K.; Nagai, Y.; Ishikura, H.; Fujii, C.; Mukaida, N. Pim-3, a proto-oncogene with serine/threonine kinase activity, is aberrantly expressed in human pancreatic cancer and phosphorylates bad to block bad-mediated apoptosis in human pancreatic cancer cell lines. Cancer Res. 2006, 66, 6741–6747. [Google Scholar] [CrossRef] [PubMed]
- Popivanova, B.K.; Ying-Yi, L.; zheng, H.; Omura, K.; Fujii, C.; Tsuneyama, K.; Mukaida, N. Proto-oncogene, Pim-3 with serine/threonine kinase activity, is aberrantly expressed in human colon cancer cells and can prevent Bad-mediated apoptosis. Cancer Sci. 2007, 98, 321–328. [Google Scholar] [CrossRef]
- Zheng, H.-C.; Tsuneyama, K.; Takahashi, H.; Miwa, S.; Sugiyama, T.; Popivanova, B.K.; Fujii, C.; Nomoto, K.; Mukaida, N.; Takano, Y. Aberrant Pim-3 expression is involved in gastric adenoma–adenocarcinoma sequence and cancer progression. J. Cancer Res. Clin. Oncol. 2007, 134, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Zhao, M.-Y.; Hei, K.-W.; Yang, B.-C.; Sun, L.; Du, X.; Li, Y.-M. Aberrant Expression of Pim-3 Promotes Proliferation and Migration of Ovarian Cancer Cells. Asian Pac. J. Cancer Prev. APJCP 2015, 16, 3325–3331. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, C.; Du, E.; Wang, A.; Yang, Y.; Guo, J.; Wang, A.; Zhang, Z.; Xu, Y. Pim-3 is a Critical Risk Factor in Development and Prognosis of Prostate Cancer. Med. Sci. Monit. 2016, 22, 4254–4260. [Google Scholar] [CrossRef]
- Aho, T.L.T.; Sandholm, J.; Peltola, K.J.; Mankonen, H.P.; Lilly, M.; Koskinen, P.J. Pim-1 kinase promotes inactivation of the pro-apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS Lett. 2004, 571, 43–49. [Google Scholar] [CrossRef]
- Yan, B.; Zemskova, M.; Holder, S.; Chin, V.; Kraft, A.; Koskinen, P.J.; Lilly, M. The PIM-2 Kinase Phosphorylates BAD on Serine 112 and Reverses BAD-induced Cell Death. J. Biol. Chem. 2003, 278, 45358–45367. [Google Scholar] [CrossRef]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef]
- Morishita, D.; Katayama, R.; Sekimizu, K.; Tsuruo, T.; Fujita, N. Pim Kinases Promote Cell Cycle Progression by Phosphorylating and Down-regulating p27Kip1 at the Transcriptional and Posttranscriptional Levels. Cancer Res. 2008, 68, 5076–5085. [Google Scholar] [CrossRef] [PubMed]
- Cen, B.; Mahajan, S.; Zemskova, M.; Beharry, Z.; Lin, Y.-W.; Cramer, S.D.; Lilly, M.B.; Kraft, A.S. Regulation of Skp2 Levels by the Pim-1 Protein Kinase[S]. J. Biol. Chem. 2010, 285, 29128–29137. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Y.; Gu, J.J.; Davitt, C.; Reeves, R.; Magnuson, N.S. Pim-2 phosphorylation of p21(Cip1/WAF1) enhances its stability and inhibits cell proliferation in HCT116 cells. Int. J. Biochem. Cell Biol. 2010, 42, 1030–1038. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Wang, Z.; Magnuson, N.S. Pim-1 Kinase-Dependent Phosphorylation of p21Cip1/WAF1 Regulates Its Stability and Cellular Localization in H1299 Cells. Mol. Cancer Res. 2007, 5, 909. [Google Scholar] [CrossRef]
- Bachmann, M.; Kosan, C.; Xing, P.X.; Montenarh, M.; Hoffmann, I.; Möröy, T. The oncogenic serine/threonine kinase Pim-1 directly phosphorylates and activates the G2/M specific phosphatase Cdc25C. Int. J. Biochem. Cell Biol. 2006, 38, 430–443. [Google Scholar] [CrossRef]
- Mochizuki, T.; Kitanaka, C.; Noguchi, K.; Muramatsu, T.; Asai, A.; Kuchino, Y. Physical and functional interactions between Pim-1 kinase and Cdc25A phosphatase. Implications for the Pim-1-mediated activation of the c-Myc signaling pathway. J. Biol. Chem. 1999, 274, 18659. [Google Scholar] [CrossRef]
- Levy, D.; Davidovich, A.; Zirkin, S.; Frug, Y.; Cohen, A.M.; Shalom, S.; Don, J. Activation of cell cycle arrest and apoptosis by the proto-oncogene Pim-2. PLoS ONE 2012, 7, e34736. [Google Scholar] [CrossRef]
- Quan, J.; Zhou, L.; Qu, J. Knockdown of Pim-3 suppresses the tumorigenicity of glioblastoma by regulating cell cycle and apoptosis. Cell. Mol. Biol. 2015, 9, 42–50. [Google Scholar]
- Bachmann, M.; Hennemann, H.; Xing, P.X.; Hoffmann, I.; Möröy, T. The oncogenic serine/threonine kinase Pim-1 phosphorylates and inhibits the activity of Cdc25C-associated kinase 1 (C-TAK1): A novel role for Pim-1 at the G2/M cell cycle checkpoint. J. Biol. Chem. 2004, 279, 48319–48328. [Google Scholar] [CrossRef]
- Zippo, A.; De Robertis, A.; Serafini, R.; Oliviero, S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat. Cell Biol. 2007, 9, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Li, X.; Magnuson, N.S. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 2008, 27, 4809–4819. [Google Scholar] [CrossRef] [PubMed]
- Beharry, Z.; Mahajan, S.; Zemskova, M.; Lin, Y.-W.; Tholanikunnel, B.G.; Xia, Z.; Smith, C.D.; Kraft, A.S. The Pim protein kinases regulate energy metabolism and cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Santio, N.M.; Eerola, S.K.; Paatero, I.; Yli-Kauhaluoma, J.; Anizon, F.; Moreau, P.; Tuomela, J.; Härkönen, P.; Koskinen, P.J. Pim Kinases Promote Migration and Metastatic Growth of Prostate Cancer Xenografts. PLoS ONE 2015, 10, e0130340. [Google Scholar] [CrossRef]
- Białopiotrowicz, E.; Górniak, P.; Noyszewska-Kania, M.; Puła, B.; Makuch-Łasica, H.; Nowak, G.; Bluszcz, A.; Szydłowski, M.; Jabłonska, E.; Piechna, K.; et al. Microenvironment-induced PIM kinases promote CXCR4-triggered mTOR pathway required for chronic lymphocytic leukaemia cell migration. J. Cell. Mol. Med. 2018, 22, 3548–3559. [Google Scholar] [CrossRef] [PubMed]
- Grundler, R.; Brault, L.; Gasser, C.; Bullock, A.N.; Dechow, T.; Woetzel, S.; Pogacic, V.; Villa, A.; Ehret, S.; Berridge, G.; et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J. Exp. Med. 2009, 206, 1957–1970. [Google Scholar] [CrossRef]
- Eerola, S.K.; Santio, N.M.; Rinne, S.; Kouvonen, P.; Corthals, G.L.; Scaravilli, M.; Scala, G.; Serra, A.; Greco, D.; Ruusuvuori, P.; et al. Phosphorylation of NFATC1 at PIM1 target sites is essential for its ability to promote prostate cancer cell migration and invasion. Cell Commun. Signal. 2019, 17, 148. [Google Scholar] [CrossRef]
- Santio, N.M.; Salmela, M.; Arola, H.; Eerola, S.K.; Heino, J.; Rainio, E.-M.; Koskinen, P.J. The PIM1 kinase promotes prostate cancer cell migration and adhesion via multiple signalling pathways. Exp. Cell Res. 2016, 342, 113–124. [Google Scholar] [CrossRef]
- Zhao, B.; Liu, L.; Mao, J.; Zhang, Z.; Wang, Q.; Li, Q. PIM1 mediates epithelial-mesenchymal transition by targeting Smads and c-Myc in the nucleus and potentiates clear-cell renal-cell carcinoma oncogenesis. Cell Death Dis. 2018, 9, 307–314. [Google Scholar] [CrossRef]
- Jie, W.; He, Q.-Y.; Luo, B.-T.; Zheng, S.-J.; Kong, Y.-Q.; Jiang, H.-G.; Li, R.-J.; Guo, J.-L.; Shen, Z.-H. Inhibition of Pim-1 attenuates the proliferation and migration in nasopharyngeal carcinoma cells. Asian Pac. J. Trop. Med. 2012, 5, 645–650. [Google Scholar] [CrossRef]
- Ren, K.; Duan, W.; Shi, Y.; Li, B.; Liu, Z.; Gong, J. Ectopic over-expression of oncogene Pim-2 induce malignant transformation of nontumorous human liver cell line L02. J. Korean Med. Sci. 2010, 25, 1017–1023. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, P.; Wang, H.; Min, X.; Wang, Y.; Tang, J.; Cheng, J.; Li, D.; Chen, X.; Cheng, F.; Wang, N.; et al. Pim-3 is expressed in endothelial cells and promotes vascular tube formation. J. Cell. Physiol. 2009, 220, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-García, M.P.; Lucena-Cacace, A.; Robles-Frías, M.J.; Narlik-Grassow, M.; Blanco-Aparicio, C.; Carnero, A. The role of PIM1/PIM2 kinases in tumors of the male reproductive system. Sci. Rep. 2016, 6, 38079. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-García, M.-P.; Lucena-Cacace, A.; Robles-Frías, M.-J.; Ferrer, I.; Narlik-Grassow, M.; Blanco-Aparicio, C.; Carnero, A. Inflammation and stem markers association to PIM1/PIM2 kinase-induced tumors in breast and uterus. Oncotarget 2017, 8, 58872–58886. [Google Scholar] [CrossRef]
- Iqbal, A.; Eckerdt, F.; Bell, J.; Nakano, I.; Giles, F.J.; Cheng, S.-Y.; Lulla, R.R.; Goldman, S.; Platanias, L.C. Targeting of glioblastoma cell lines and glioma stem cells by combined PIM kinase and PI3K-p110α inhibition. Oncotarget 2016, 7, 33192–33201. [Google Scholar] [CrossRef]
- Seifert, C.; Balz, E.; Herzog, S.; Korolev, A.; Gaßmann, S.; Paland, H.; Fink, M.A.; Grube, M.; Marx, S.; Jedlitschky, G.; et al. PIM1 Inhibition Affects Glioblastoma Stem Cell Behavior and Kills Glioblastoma Stem-like Cells. Int. J. Mol. Sci. 2021, 22, 11126. [Google Scholar] [CrossRef]
- Xie, Y.; Bayakhmetov, S. PIM1 kinase as a promise of targeted therapy in prostate cancer stem cells. Mol. Clin. Oncol. 2016, 4, 13–17. [Google Scholar] [CrossRef][Green Version]
- Li, T.; Wang, Z.; Hou, Y.-F.; Li, Y.-Y. Pim-3 Regulates Stemness of Pancreatic Cancer Cells via Activating STAT3 Signaling Pathway. J. Cancer 2017, 8, 1530–1541. [Google Scholar] [CrossRef]
- Xu, D.; Cobb, M.G.; Gavilano, L.; Witherspoon, S.M.; Williams, D.; White, C.D.; Taverna, P.; Bednarski, B.K.; Kim, H.J.; Baldwin, A.S.; et al. Inhibition of oncogenic Pim-3 kinase modulates transformed growth and chemosensitizes pancreatic cancer cells to gemcitabine. Cancer Biol. Ther. 2013, 14, 492–501. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Wang, Z.; Li, B.; Zhang, Y.-J.; Li, Y.-Y. Pim-3 contributes to radioresistance through regulation of the cell cycle and DNA damage repair in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2016, 473, 296–302. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Wang, P.; Yang, C.; Li, S. Reduced pim-1 expression increases chemotherapeutic drug sensitivity in human androgen-independent prostate cancer cells by inducing apoptosis. Exp. Ther. Med. 2019, 18, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xu, K.; Linn, D.E.; Yang, X.; Guo, Z.; Shimelis, H.; Nakanishi, T.; Ross, D.D.; Chen, H.; Fazli, L.; et al. The 44-kDa Pim-1 Kinase Phosphorylates BCRP/ABCG2 and Thereby Promotes Its Multimerization and Drug-resistant Activity in Human Prostate Cancer Cells. J. Biol. Chem. 2008, 283, 3349–3356. [Google Scholar] [CrossRef] [PubMed]
- Musiani, D.; Hammond, D.E.; Cirillo, L.; Erriquez, J.; Olivero, M.; Clague, M.J.; Di Renzo, M.F. PIM2 Kinase Is Induced by Cisplatin in Ovarian Cancer Cells and Limits Drug Efficacy. J. Proteome Res. 2014, 13, 4970–4982. [Google Scholar] [CrossRef]
- Guo, H.; Dong, J.; Hu, S.; Cai, X.; Tang, G.; Dou, J.; Tian, M.; He, F.; Nie, Y.; Fan, D. Biased random walk model for the prioritization of drug resistance associated proteins. Sci. Rep. 2015, 5, 10857. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Sui, Z.-G.; Xu, W.; Quan, X.-H.; Sun, J.-L.; Li, X.; Ji, H.-Y.; Jing, F.-B. Ubenimex suppresses Pim-3 kinase expression by targeting CD13 to reverse MDR in HCC cells. Oncotarget 2017, 8, 72652–72665. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Padi, S.K.R.; Bearss, J.J.; Pandey, R.; Okumura, K.; Beltran, H.; Song, J.H.; Kraft, A.S.; Olive, V. PIM protein kinases regulate the level of the long noncoding RNA H19 to control stem cell gene transcription and modulate tumor growth. Mol. Oncol. 2020, 14, 974–990. [Google Scholar] [CrossRef]
- Brumbaugh, J.; Hou, Z.; Russell, J.D.; Howden, S.E.; Yu, P.; Ledvina, A.R.; Coon, J.J.; Thomson, J.A. Phosphorylation regulates human OCT4. Proc. Natl. Acad. Sci. USA 2012, 109, 7162–7168. [Google Scholar] [CrossRef]
- Chikazawa, N.; Tanaka, H.; Tasaka, T.; Nakamura, M.; Tanaka, M.; Onishi, H.; Katano, M. Inhibition of Wnt signaling pathway decreases chemotherapy-resistant side-population colon cancer cells. Anticancer Res. 2010, 30, 2041–2048. [Google Scholar]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Investig. 2011, 121, 3804–3809. [Google Scholar] [CrossRef]
- Stafman, L.L.; Williams, A.P.; Garner, E.F.; Aye, J.M.; Stewart, J.E.; Yoon, K.J.; Whelan, K.; Beierle, E.A. Targeting PIM Kinases Affects Maintenance of CD133 Tumor Cell Population in Hepatoblastoma. Transl. Oncol. 2019, 12, 200–208. [Google Scholar] [CrossRef]
- Stafman, L.L.; Waldrop, M.G.; Williams, A.P.; Aye, J.M.; Stewart, J.E.; Mroczek-Musulman, E.; Yoon, K.J.; Whelan, K.; Beierle, E.A. The presence of PIM3 increases hepatoblastoma tumorigenesis and tumor initiating cell phenotype and is associated with decreased patient survival. J. Pediatric Surg. 2019, 54, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Wadhwani, N.; Markert, H.R.; Marayati, R.; Bownes, L.V.; Quinn, C.H.; Aye, J.M.; Stewart, J.E.; Yoon, K.J.; Beierle, E.A. PIM447 inhibits oncogenesis and potentiates cisplatin effects in hepatoblastoma. J. Pediatric Surg. 2021, 56, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Marayati, R.; Stafman, L.L.; Williams, A.P.; Bownes, L.V.; Quinn, C.H.; Markert, H.R.; Easlick, J.L.; Stewart, J.E.; Crossman, D.K.; Mroczek-Musulman, E.; et al. CRISPR/Cas9-mediated knockout of PIM3 suppresses tumorigenesis and cancer cell stemness in human hepatoblastoma cells. Cancer Gene Ther. 2021, 29, 558–572. [Google Scholar] [CrossRef]
- Marayati, R.; Stafman, L.L.; Williams, A.P.; Bownes, L.V.; Quinn, C.H.; Aye, J.M.; Stewart, J.E.; Yoon, K.J.; Anderson, J.C.; Willey, C.D.; et al. PIM kinases mediate resistance to cisplatin chemotherapy in hepatoblastoma. Sci. Rep. 2021, 11, 5914–5984. [Google Scholar] [CrossRef]
- Brunen, D.; de Vries, R.C.; Lieftink, C.; Beijersbergen, R.L.; Bernards, R. PIM Kinases Are a Potential Prognostic Biomarker and Therapeutic Target in Neuroblastoma. Mol. Cancer Ther. 2018, 17, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Trigg, R.M.; Turner, S.D. ALK in Neuroblastoma: Biological and Therapeutic Implications. Cancers 2018, 10, 113. [Google Scholar] [CrossRef]
- Mohlin, S.; Hansson, K.; Radke, K.; Martinez, S.; Blanco-Aparicio, C.; Garcia-Ruiz, C.; Welinder, C.; Esfandyari, J.; O’Neill, M.; Pastor, J.; et al. Anti-tumor effects of PIM/PI 3K/mTOR triple kinase inhibitor IBL-302 in neuroblastoma. EMBO Mol. Med. 2020, 12, e11749. [Google Scholar] [CrossRef]
- Liao, Y.; Feng, Y.; Shen, J.; Gao, Y.; Cote, G.; Choy, E.; Harmon, D.; Mankin, H.; Hornicek, F.; Duan, Z. Clinical and biological significance of PIM1 kinase in osteosarcoma. J. Orthop. Res. 2016, 34, 1185–1194. [Google Scholar] [CrossRef]
- Narlik-Grassow, M.; Blanco-Aparicio, C.; Cecilia, Y.; Peregrina, S.; Garcia-Serelde, B.; Munoz-Galvan, S.; Canamero, M.; Carnero, A. The essential role of PIM kinases in sarcoma growth and bone invasion. Carcinogenesis 2012, 33, 1479–1486. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, J.; Xing, C.; Wei, S.; Guo, N.; Wang, Y. miR-486 inhibited osteosarcoma cells invasion and epithelial-mesenchymal transition by targeting PIM1. Cancer Biomark. 2018, 33, 269–277. [Google Scholar] [CrossRef]
- Xia, Z.; Knaak, C.; Ma, J.; Beharry, Z.M.; McInnes, C.; Wang, W.; Kraft, A.S.; Smith, C.D. Synthesis and Evaluation of Novel Inhibitors of Pim-1 and Pim-2 Protein Kinases. J. Med. Chem. 2009, 52, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Hasegawa, T.; Kojima, H.; Okabe, T.; Nagano, T. Design and Synthesis of Potent and Selective PIM Kinase Inhibitors by Targeting Unique Structure of ATP-Binding Pocket. ACS Med. Chem. Lett. 2017, 8, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Jeyapal, G.P.; Chandrasekar, M.J.N.; Krishnasamy, R.; Selvaraj, J.; Mohammad, M.; Nanjan, M. Potential pharmacological inhibitors of PIM kinase under clinical trials. Anticancer Agents Med. Chem. 2018, 8, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Haddach, M.; Pierre, F.; Ryckman, D.M. Potential use of selective and nonselective Pim kinase inhibitors for cancer therapy. J. Med. Chem. 2012, 55, 8199–8208. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.D.; Langowski, J.L.; Niu, X.-H.; Basham, S.; Chan, J.; Jianjun, Y.U.; Doyle, M.; Feucht, P.; Warne, R.; Narberes, J.; et al. Pan-PIM Kinase Inhibition Provides a Novel Therapy for Treating Hematologic Cancers. Clin. Cancer Res. 2014, 20, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Blackaby, W.; Allen, V.; Chan, G.K.Y.; Chang, J.H.; Chiang, P.-C.; Diène, C.; Drummond, J.; Do, S.; Fan, E.; et al. Optimization of Pan-Pim Kinase Activity and Oral Bioavailability Leading to Diaminopyrazole (GDC-0339) for the Treatment of Multiple Myeloma. J. Med. Chem. 2019, 62, 2140–2153. [Google Scholar] [CrossRef]
- Wang, X.; Kolesnikov, A.; Tay, S.; Chan, G.; Chao, Q.; Do, S.; Drummond, J.; Ebens, A.J.; Liu, N.; Ly, J.; et al. Discovery of 5-Azaindazole (GNE-955) as a Potent Pan-Pim Inhibitor with Optimized Bioavailability. J. Med. Chem. 2017, 60, 4458–4473. [Google Scholar] [CrossRef]
- Keeton, E.K.; McEachern, K.; Dillman, K.S.; Palakurthi, S.; Cao, Y.; Grondine, M.R.; Kaur, S.; Wang, S.; Chen, Y.; Wu, A.; et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 2014, 123, 905–913. [Google Scholar] [CrossRef]
- Burger, M.T.; Nishiguchi, G.; Han, W.; Lan, J.; Simmons, R.; Atallah, G.; Ding, Y.; Tamez, V.; Zhang, Y.; Mathur, M.; et al. Identification of N-(4-((1R,3S,5S)-3-Amino-5-methylcyclohexyl)yridine-3-yl)-6-(2,6-difluorophenyl)-5-fluoropicolinamide (PIM447), a Potent and Selective Proviral Insertion Site of Moloney Murine Leukemia (PIM) 1, 2, and 3 Kinase Inhibitor in Clinical Trials for Hematological Malignancies. J. Med. Chem. 2015, 58, 8373–8386. [Google Scholar] [CrossRef]
- Solomon, S.R.; Montesinos, P.; Nazha, A.; Strickland, S.A.; Martinelli, G.; Santoro, A.; Walter, R.B.; Cook, R.J.; Calbacho, M.; Vives, S.; et al. Updated results from DIAMOND-01 (CLI24-001) trial: A phase I/II study of SEL24/MEN1703, a first-in-class dual PIM/FLT3 kinase inhibitor, in acute myeloid leukemia. J. Clin. Oncol. 2021, 39, 7023. [Google Scholar] [CrossRef]
- Kaewchim, K.; Glab-Ampai, K.; Mahasongkram, K.; Chulanetra, M.; Seesuay, W.; Chaicumpa, W.; Sookrung, N. Engineered Fully Human Single-Chain Monoclonal Antibodies to PIM2 Kinase. Molecules 2021, 26, 6436. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, Y.; Shi, S.; Li, K.; Zhang, L.; Gao, J. Insights into the Interaction Mechanisms of the Proviral Integration Site of Moloney Murine Leukemia Virus (Pim) Kinases with Pan-Pim Inhibitors PIM447 and AZD1208: A Molecular Dynamics Simulation and MM/GBSA Calculation Study. Int. J. Mol. Sci. 2019, 20, 5410. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Thomas, S.K.; Ocio, E.M.; Guenther, A. Yeow-Tee, G.; Talpaz, M.; Hohmann, N.; Zhao, S.; Xiang, F.; Simon, C.; et al. The first-in-human study of the pan-PIM kinase inhibitor PIM447 in patients with relapsed and/or refractory multiple myeloma. Leukemia 2019, 33, 2924–2933. [Google Scholar] [CrossRef] [PubMed]
- Iida, S.; Sunami, K.; Minami, H.; Hatake, K.; Sekiguchi, R.; Natsume, K.; Ishikawa, N.; Rinne, M.; Taniwaki, M. A phase I, dose-escalation study of oral PIM447 in Japanese patients with relapsed and/or refractory multiple myeloma. Int. J. Hematol. 2021, 113, 797–806. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Available online: ClinicalTrials.gov (accessed on 16 June 2022).
- Martinelli, G.; Santoro, A.; Gambacorti-Passerini, C.; Polo, S.V.; Solomon, S.R.; Mukherjee, S.; Levy, M.Y.; Wierzbowska, A.; Calbacho, M.; Angelosanto, N.; et al. Phase 1/2 study of SEL24/MEN1703, a first-in-class dual PIM/FLT3 kinase inhibitor, in patients with IDH1/2-mutated acute myeloid leukemia: The DIAMOND-01 trial. J. Clin. Oncol. 2022, 40, 7024. [Google Scholar] [CrossRef]
- Solomon, S.R.; Nazha, A.; Strickland, S.A.; Walter, R.B.; Valimberti, I.; Tagliavini, A.; Mazzei, P.; Fiesoli, C.; Scartoni, S.; Bellarosa, D.; et al. First in Human Study of SEL24/MEN1703, First in Class, Orally Available Dual PIM/FLT3 Kinase Inhibitor, in Patients with Acute Myeloid Leukemia. Blood 2019, 134, 3920. [Google Scholar] [CrossRef]
- Paoli, A.; Bellarosa, D.; Bressan, A.; Bertolini, F.; Solomon, S.R.; Montesinos, P.; Mukherjee, S.; Strickland, S.A.; Marconi, G.; Santoro, A.; et al. SEL24/MEN1703 Inhibits PIM/FLT3 Downstream Target in Acute Myeloid Leukemia (AML) Patients: Results of the Pharmacodynamics (PD) Assay and Genomic Profiling in the First-in-Human Diamond-01 Trial. Blood 2021, 138, 3436. [Google Scholar] [CrossRef]


{kind=link}
| Pharmacologic Agent | Cancer Type or Condition | Clinical Trial Phases Conducted | Clinical Trial Number(s) |
|---|---|---|---|
| AZD1208 | Acute myelogenous leukemia | Phase I (Terminated) | NCT01489722 |
| Advanced solid tumors, malignant lymphoma | Phase I (Completed) | NCT01588548 | |
| PIM447 | Myelofibrosis | Phase I (Completed) | NCT02370706 |
| Multiple myeloma | Phase I (Completed) | NCT02160951 | |
| Relapsed and/or refractory multiple myeloma | Phase I (Completed) | NCT02144038 | |
| Acute myeloid leukemia, high risk myelodysplastic syndrome | Phase I (Completed) | NCT02078609 | |
| Relapsed and/or refractory multiple myeloma | Phase I (Completed) | NCT01456689 | |
| SGI-1776 | Relapsed and/or refractory leukemias | Phase I (Withdrawn) | NCT01239108 |
| Prostate cancer, Non-Hodgkin lymphoma | Phase I (Terminated) | NCT00848601 | |
| INCB053914 | Relapsed and/or refractory multiple myeloma | Phase I (Withdrawn) | NCT04355039 |
| Advanced solid tumors | Phase I/II (Terminated) | NCT02587598 | |
| Relapsed and/or refractory diffuse large B cell lymphoma | Phase I (Completed) | NCT03688152 | |
| CX-4945 | Cholangiocarcinoma | Phase I/II (Completed) | NCT02128282 |
| Multiple myeloma | Phase I (Unknown) | NCT01199718 | |
| Advanced solid tumors, breast cancer, inflammatory breast cancer, Castleman disease, multiple myeloma | Phase I (Unknown) | NCT00891280 | |
| SEL24/MEN1703 | Acute myeloid leukemia | Phase I/II (Recruiting) | NCT03008187 |
| LY-2835219 (Abemaciclib) | Relapsed or refractory pediatric solid tumors | Phase I (Recruiting) | NCT04238819, NCT02644460 |
| Pediatric brain tumors | Phase I (Recruiting) | NCT02644460 | |
| Advanced malignancies | Phase I (Active, Completed) | NCT02117648, NCT01394016, NCT02919696, NCT04071262, NCT02857270, NCT01655225, NCT05307705, NCT02791334, NCT02745769 | |
| Breast cancer, metastatic breast cancer | Phase I (Active, Completed), Phase II (Active, Completed, Recruiting), Phase III (Active, Completed, Recruiting), Phase IV (Recruiting, Terminated, Withdrawn) | NCT02831530, NCT02441946, NCT02246621, NCT02102490, NCT05169567, NCT02763566, NCT04752332, NCT03988114, NCT02057133, NCT03703466, NCT03763604, NCT02779751, NCT02675231, NCT02792725, NCT03155997, NCT02747004, NCT04031885, NCT02107703, NCT03130439, NCT04707196, NCT05169567, NCT04975308, NCT04188548, NCT02784795, NCT02688088, NCT04305834, NCT03979508, NCT05307705, NCT04481113, NCT04351230, NCT04256941, NCT03878524 | |
| Non-small cell lung cancer, metastatic non-small cell lung cancer | Phase I (Active, Completed), Phase II (Completed), Phase III (Active) | NCT02079636, NCT02450539, NCT02779751, NCT02152631, NCT02411591 | |
| Sarcoma, dedifferentiated liposarcoma | Phase II (Active), Phase III (Recruiting) | NCT02846987, NCT04967521 | |
| Brain tumor, recurrent glioblastoma | Phase II (Active, Withdrawn) | NCT03220646, NCT02981940, NCT04118036 | |
| Metastatic castration-resistant prostate cancer | Phase II (Active, Recruiting), Phase III (Recruiting) | NCT04408924, NCT03706365, NCT05288166 | |
| Mantle cell lymphoma | Phase II (Active) | NCT01739309 | |
| Pancreatic ductal adenocarcinoma | Phase II (Completed) | NCT02981342 | |
| Metastatic breast or non-small cell lung cancer, or melanoma with brain metastasis | Phase II (Completed, Withdrawn) | NCT02308020, NCT04585724 | |
| Small cell lung cancer | Phase I (Recruiting) | NCT04010357 | |
| Metastatic cancer, BRAF V600E, MEK1, MEK2, ERK, KRAS, or RAF1 gene mutations | Phase II (Recruiting) | NCT04534283 | |
| Endometrial cancer, metastatic endometrial cancer | Phase I (Recruiting), Phase II (Active) | NCT04188548, NCT04049227, NCT04469764 | |
| Metastatic or locally advanced anaplastic/undifferentiated thyroid cancer | Phase II (Recruiting) | NCT04552769 | |
| Unresectable of metastatic colorectal cancer | Phase I/II (Recruiting) | NCT04616183 | |
| Mesothelioma | Phase II (Recruiting) | NCT03654833 | |
| Multiple myeloma | Phase I/II (Recruiting) | NCT03732703 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Julson, J.R.; Marayati, R.; Beierle, E.A.; Stafman, L.L. The Role of PIM Kinases in Pediatric Solid Tumors. Cancers 2022, 14, 3565. https://doi.org/10.3390/cancers14153565
Julson JR, Marayati R, Beierle EA, Stafman LL. The Role of PIM Kinases in Pediatric Solid Tumors. Cancers. 2022; 14(15):3565. https://doi.org/10.3390/cancers14153565
Chicago/Turabian StyleJulson, Janet Rae, Raoud Marayati, Elizabeth Ann Beierle, and Laura Lee Stafman. 2022. "The Role of PIM Kinases in Pediatric Solid Tumors" Cancers 14, no. 15: 3565. https://doi.org/10.3390/cancers14153565
APA StyleJulson, J. R., Marayati, R., Beierle, E. A., & Stafman, L. L. (2022). The Role of PIM Kinases in Pediatric Solid Tumors. Cancers, 14(15), 3565. https://doi.org/10.3390/cancers14153565