
Identification of Malignant Cell Populations Associated with Poor Prognosis in High-Grade Serous Ovarian Cancer Using Single-Cell RNA Sequencing

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Construction of scRNA-seq and RNA-seq Libraries from Ovarian Cancer Specimens
2.2. Bioinformatics
2.2.1. scRNA-seq Data Analysis
2.2.2. Bulk RNA-seq Data Analysis
2.2.3. Digital Cytometry Analysis
2.2.4. Copy Number Variation (CNV) Analysis
2.2.5. Single-Cell Trajectory Analysis
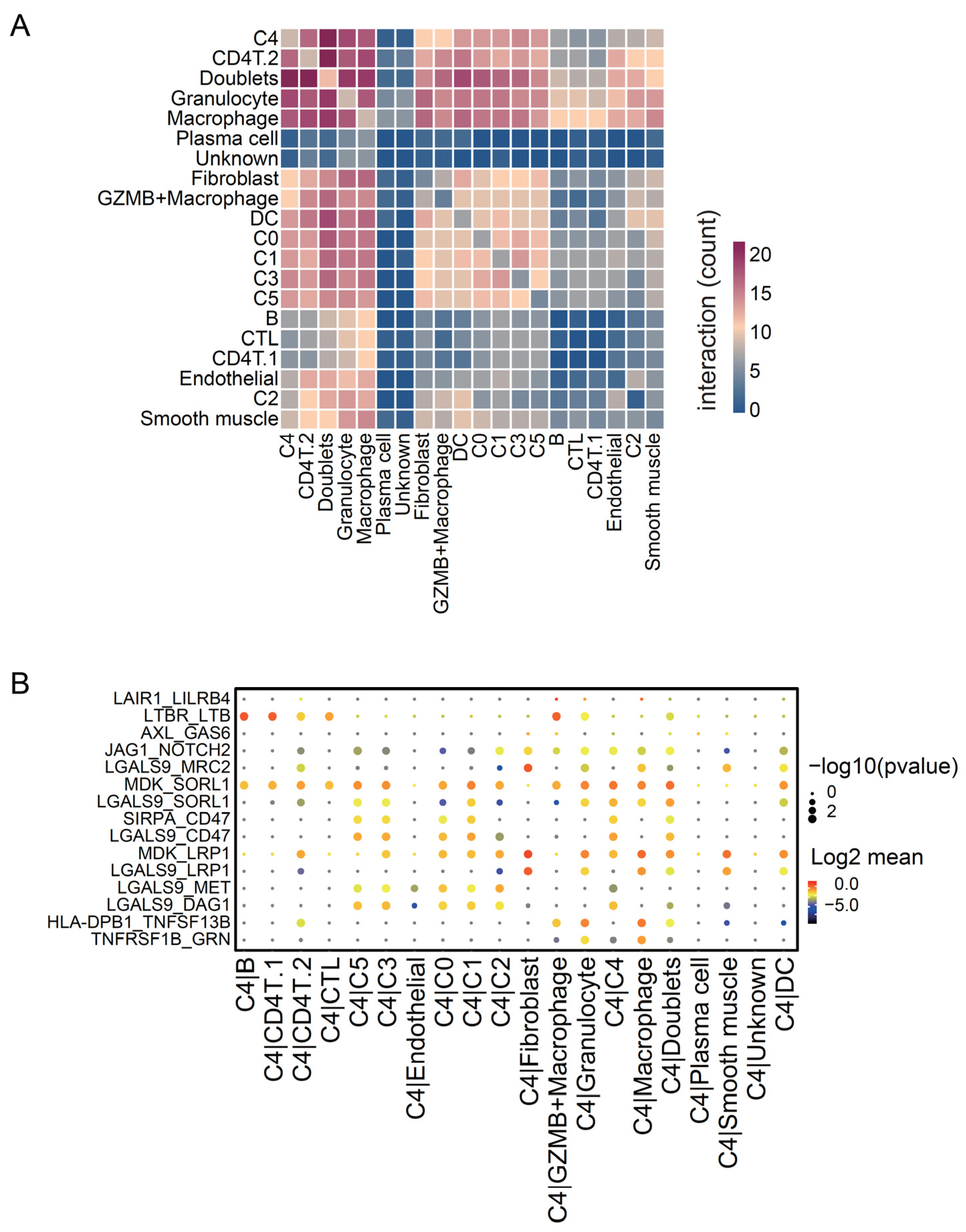
2.2.6. Cell–Cell Interaction Analysis
3. Results
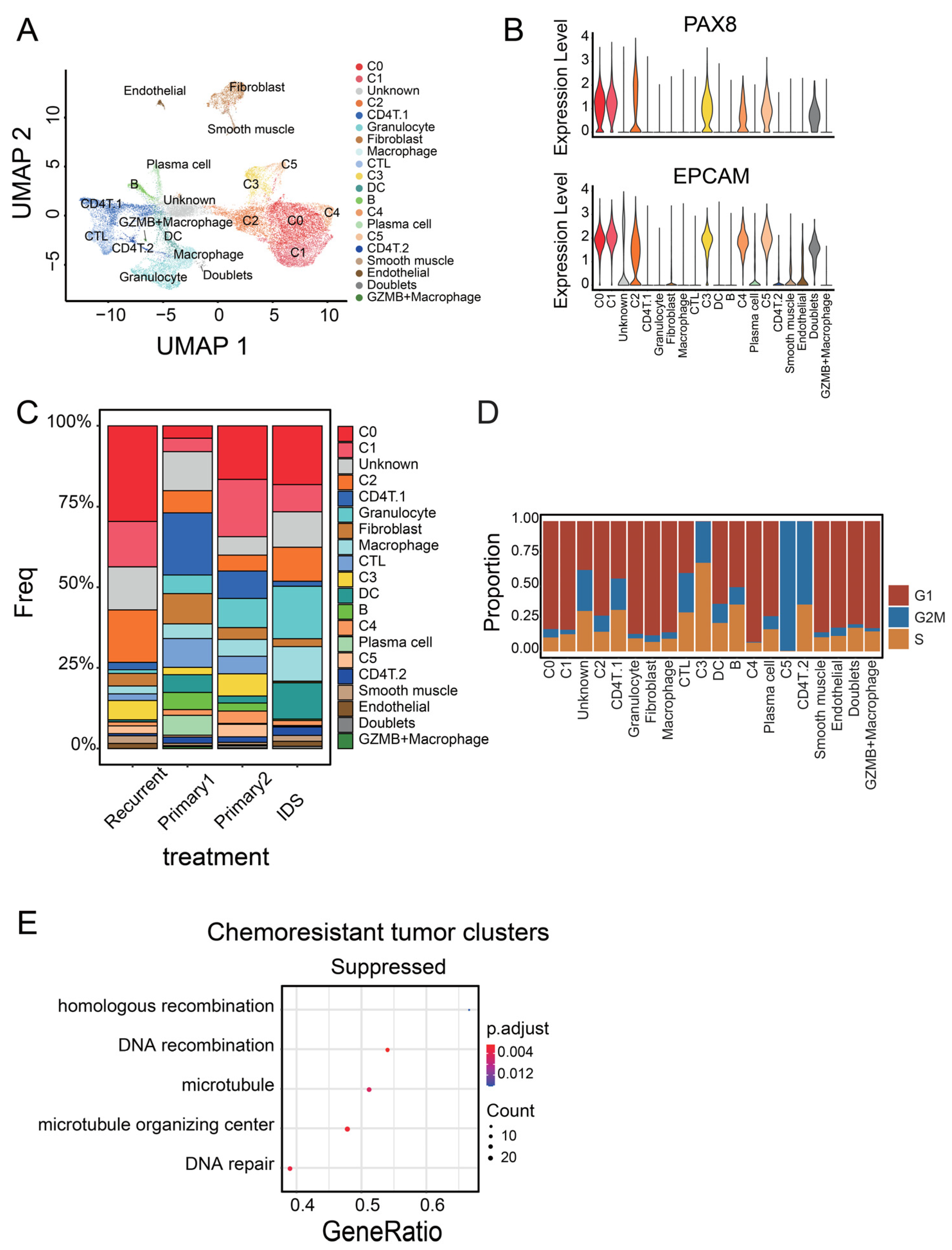
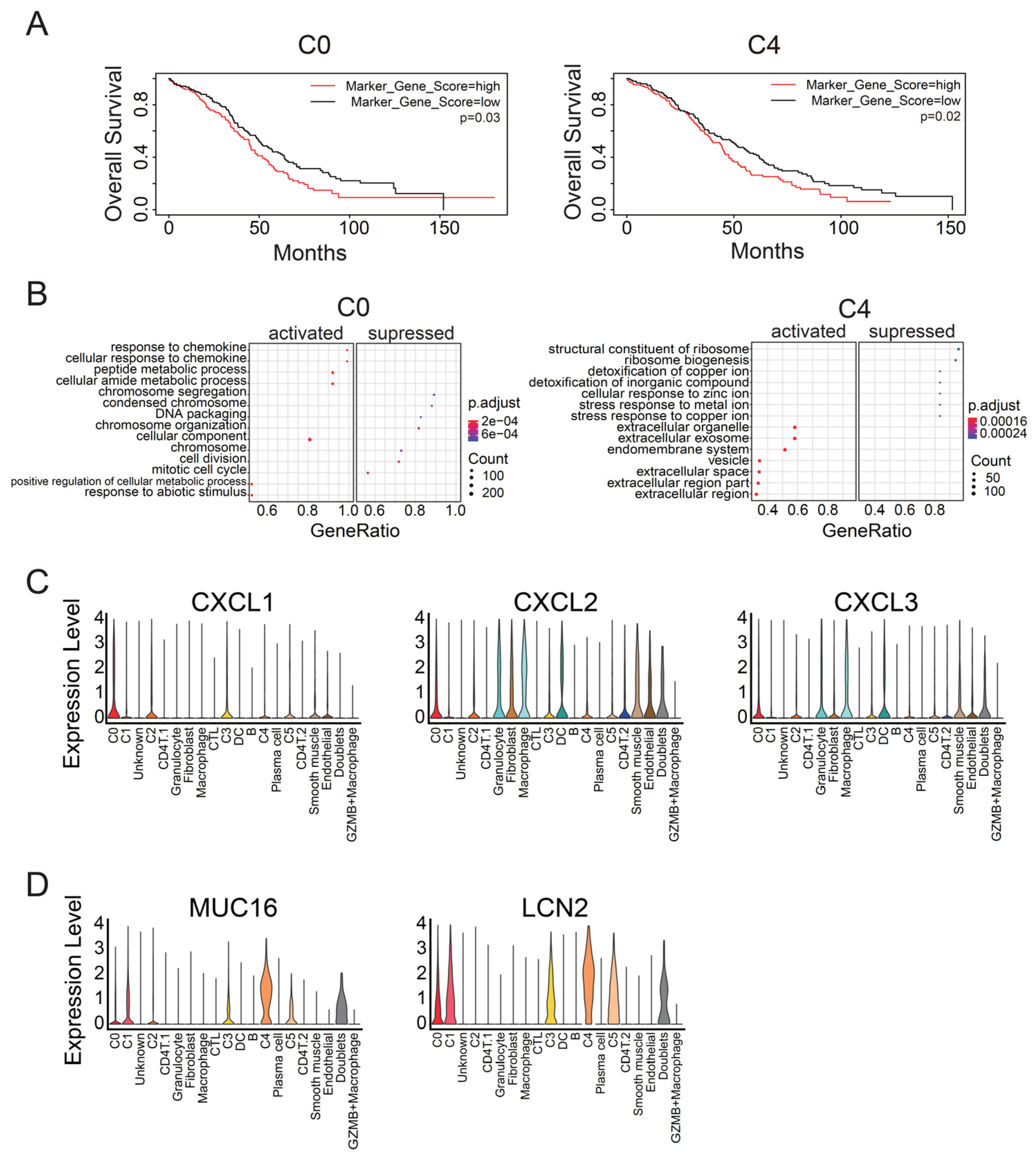
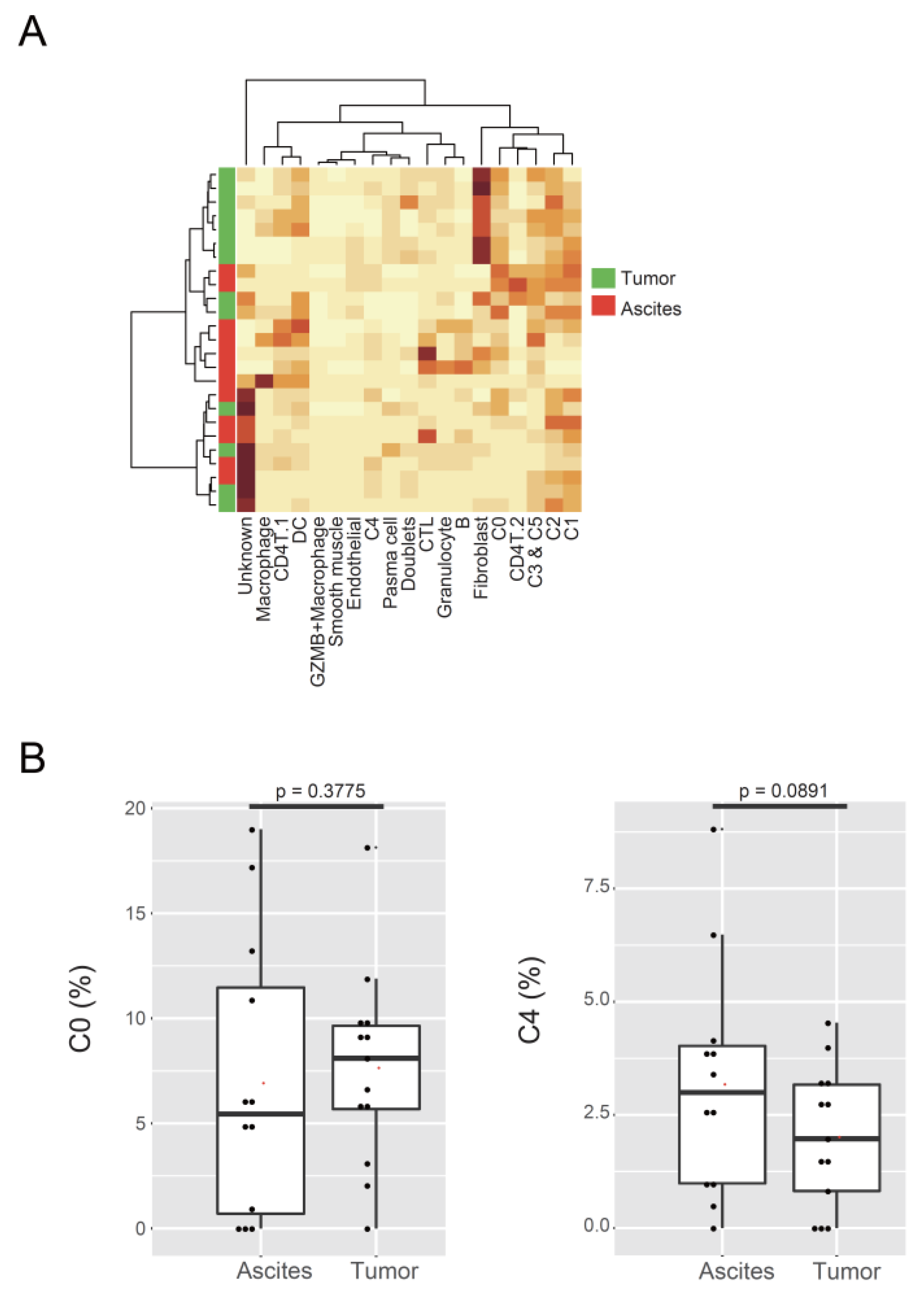
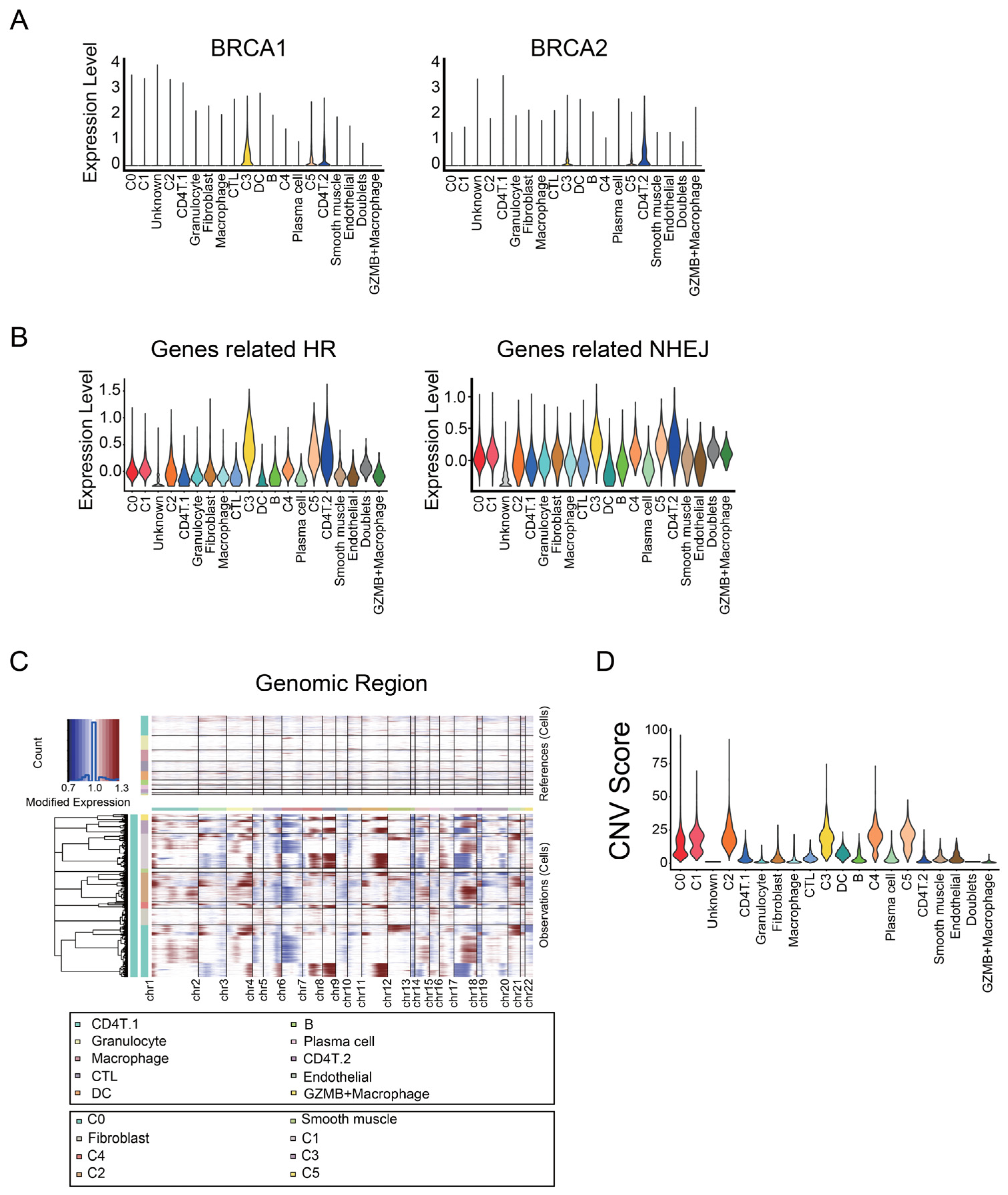
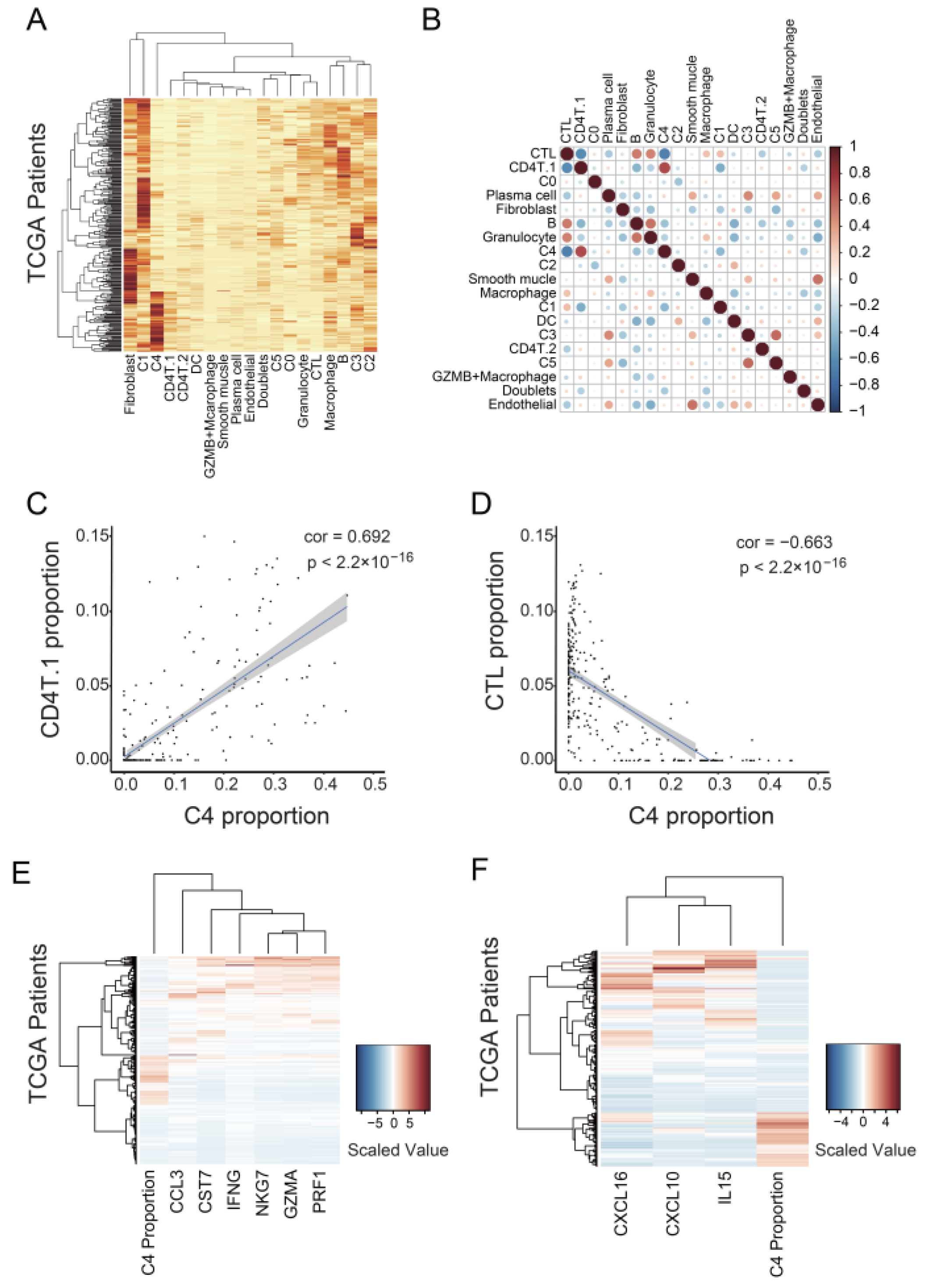
3.1. Identification of Malignant Tumor Clusters Associated with Poor Prognosis
3.2. Identifying Malignant Tumor Cluster Properties
3.3. C0 Cluster Includes the Oldest Epithelial Cancer Cells and Cancer Stem-Like Cells
3.4. C4 Cluster Is Associated with Decreasing Tumor-Infiltrating CTL Population
4. Discussion
4.1. Identification of Marker Genes for Cancer Stem-Like Cells in Ovarian Cancer
4.2. C4 Cluster Is a New Subtype Associated with Poor Prognosis in Ovarian Cancer
4.3. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial Ovarian Cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef] [Green Version]
- Rossing, M.A.; Wicklund, K.G.; Cushing-Haugen, K.L.; Weiss, N.S. Predictive Value of Symptoms for Early Detection of Ovarian Cancer. JNCI J. Natl. Cancer Inst. 2010, 102, 222–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US Preventive Services Task Force; Grossman, D.C.; Curry, S.J.; Owens, D.K.; Barry, M.J.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W.; Kemper, A.R.; Krist, A.H.; et al. Screening for Ovarian Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 319, 588. [Google Scholar] [CrossRef] [Green Version]
- Hellstrom, I.; Raycraft, J.; Hayden-Ledbetter, M.; Ledbetter, J.A.; Schummer, M.; McIntosh, M.; Drescher, C.; Urban, N.; Hellstrom, K.E. The HE4 (WFDC2) Protein Is a Biomarker for Ovarian Carcinoma. Cancer Res. 2003, 63, 3695–3700. [Google Scholar]
- Yang, W.-L.; Lu, Z.; Bast, R.C. The Role of Biomarkers in the Management of Epithelial Ovarian Cancer. Expert Rev. Mol. Diagn. 2017, 17, 577–591. [Google Scholar] [CrossRef]
- Buys, S.S. Effect of Screening on Ovarian Cancer Mortality: The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 2011, 305, 2295. [Google Scholar] [CrossRef]
- Ozga, M.; Aghajanian, C.; Myers-Virtue, S.; McDonnell, G.; Jhanwar, S.; Hichenberg, S.; Sulimanoff, I. A Systematic Review of Ovarian Cancer and Fear of Recurrence. Palliat. Support. Care 2015, 13, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial Ovarian Cancer: Evolution of Management in the Era of Precision Medicine. CA A Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [Green Version]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian Cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Swanton, C. Intratumor Heterogeneity: Evolution through Space and Time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Han, Y.; Kim, S.I.; Kim, H.-S.; Kim, S.J.; Song, Y.S. Tumor Evolution and Chemoresistance in Ovarian Cancer. NPJ Precis. Oncol. 2018, 2, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Sánchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 2017, 170, 927–938.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, A.J.; Menzin, A.; Whyte, J.; Lovecchio, J.; Liew, A.; Khalili, H.; Bhuiya, T.; Gregersen, P.K.; Lee, A.T. Identification of Grade and Origin Specific Cell Populations in Serous Epithelial Ovarian Cancer by Single Cell RNA-Seq. PLoS ONE 2018, 13, e0206785. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Z.; Gao, Y.; Li, J.; Zhao, X.; Yue, W. Single-Cell RNA-Sequencing Portraying Functional Diversity and Clinical Implications of IFI6 in Ovarian Cancer. Front. Cell Dev. Biol. 2021, 9, 677697. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Wang, F.; Gao, C.; Cao, Y.; Wang, J. Identification of Specific Cell Subpopulations and Marker Genes in Ovarian Cancer Using Single-Cell RNA Sequencing. BioMed Res. Int. 2021, 2021, 1005793. [Google Scholar] [CrossRef]
- Kawamura, N.; Nimura, K.; Saga, K.; Ishibashi, A.; Kitamura, K.; Nagano, H.; Yoshikawa, Y.; Ishida, K.; Nonomura, N.; Arisawa, M.; et al. SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression. Cancer Res. 2019, 79, 5204–5217. [Google Scholar] [CrossRef] [Green Version]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring Cell–Cell Communication from Combined Expression of Multi-Subunit Ligand–Receptor Complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.-E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-Cell Reconstruction of the Early Maternal–Fetal Interface in Humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed Graph Embedding Resolves Complex Single-Cell Trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The Dynamics and Regulators of Cell Fate Decisions Are Revealed by Pseudotemporal Ordering of Single Cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The Single-Cell Transcriptional Landscape of Mammalian Organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019, 8, 281–291.e9. [Google Scholar] [CrossRef] [Green Version]
- Bergen, V.; Lange, M.; Peidli, S.; Wolf, F.A.; Theis, F.J. Generalizing RNA Velocity to Transient Cell States through Dynamical Modeling. Nat. Biotechnol. 2020, 38, 1408–1414. [Google Scholar] [CrossRef]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lönnerberg, P.; Furlan, A.; et al. RNA Velocity of Single Cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef] [Green Version]
- Bergen, V.; Soldatov, R.A.; Kharchenko, P.V.; Theis, F.J. RNA Velocity—Current Challenges and Future Perspectives. Mol. Syst. Biol. 2021, 17, e10282. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial Reconstruction of Single-Cell Gene Expression Data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [Green Version]
- Tangjitgamol, S.; Manusirivithaya, S.; Laopaiboon, M.; Lumbiganon, P.; Bryant, A. Interval Debulking Surgery for Advanced Epithelial Ovarian Cancer: A Cochrane systematic review. Gynecol. Oncol. 2009, 112, 257–264. [Google Scholar] [CrossRef]
- Laury, A.R.; Perets, R.; Piao, H.; Krane, J.F.; Barletta, J.A.; French, C.; Chirieac, L.R.; Lis, R.; Loda, M.; Hornick, J.L.; et al. A Comprehensive Analysis of PAX8 Expression in Human Epithelial Tumors. Am. J. Surg. Pathol. 2011, 35, 816–826. [Google Scholar] [CrossRef] [Green Version]
- Trzpis, M.; McLaughlin, P.M.J.; de Leij, L.M.F.H.; Harmsen, M.C. Epithelial Cell Adhesion Molecule. Am. J. Pathol. 2007, 171, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Verdier-Pinard, P.; Fernandez-Fuentes, N.; Burd, B.; Angeletti, R.; Fiser, A.; Horwitz, S.B.; Orr, G.A. Insights into the Mechanism of Microtubule Stabilization by Taxol. Proc. Natl. Acad. Sci. USA 2006, 103, 10166–10173. [Google Scholar] [CrossRef] [Green Version]
- Brabec, V.; Kasparkova, J. Modifications of DNA by Platinum Complexes. Drug Resist. Updates 2005, 8, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In Search of Definitions: Cancer-associated Fibroblasts and Their Markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izar, B.; Tirosh, I.; Stover, E.H.; Wakiro, I.; Cuoco, M.S.; Alter, I.; Rodman, C.; Leeson, R.; Su, M.-J.; Shah, P.; et al. A Single-Cell Landscape of High-Grade Serous Ovarian Cancer. Nat. Med. 2020, 26, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Giamougiannis, P.; Martin-Hirsch, P.L.; Martin, F.L. The Evolving Role of MUC16 (CA125) in the Transformation of Ovarian Cells and the Progression of Neoplasia. Carcinogenesis 2021, 42, 327–343. [Google Scholar] [CrossRef] [PubMed]
- Gubbels, J.A.; Belisle, J.; Onda, M.; Rancourt, C.; Migneault, M.; Ho, M.; Bera, T.K.; Connor, J.; Sathyanarayana, B.K.; Lee, B.; et al. Mesothelin-MUC16 Binding Is a High Affinity, N-Glycan Dependent Interaction That Facilitates Peritoneal Metastasis of Ovarian Tumors. Mol. Cancer 2006, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Graf, R.P.; Eskander, R.; Brueggeman, L.; Stupack, D.G. Association of Copy Number Variation Signature and Survival in Patients with Serous Ovarian Cancer. JAMA Netw. Open 2021, 4, e2114162. [Google Scholar] [CrossRef]
- Mihanfar, A.; Aghazadeh Attari, J.; Mohebbi, I.; Majidinia, M.; Kaviani, M.; Yousefi, M.; Yousefi, B. Ovarian Cancer Stem Cell: A Potential Therapeutic Target for Overcoming Multidrug Resistance. J. Cell. Physiol. 2019, 234, 3238–3253. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ Cytotoxic T Lymphocytes in Cancer Immunotherapy: A Review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ Tumor-Infiltrating Lymphocytes and a High CD8+/Regulatory T Cell Ratio Are Associated with Favorable Prognosis in Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [Green Version]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the Cancer Microenvironment and Their Relevance in Cancer Immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Matloubian, M.; David, A.; Engel, S.; Ryan, J.E.; Cyster, J.G. A Transmembrane CXC Chemokine Is a Ligand for HIV-Coreceptor Bonzo. Nat. Immunol. 2000, 1, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Budagian, V.; Bulanova, E.; Paus, R.; Bulfonepaus, S. IL-15/IL-15 Receptor Biology: A Guided Tour through an Expanding Universe. Cytokine Growth Factor Rev. 2006, 17, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Caligiuri, M.A. Interleukin 15: Biology and Relevance to Human Disease. Blood 2001, 97, 14–32. [Google Scholar] [CrossRef]
- Vaitaitis, G.M.; Wagner, D.H. Galectin-9 Controls CD40 Signaling through a Tim-3 Independent Mechanism and Redirects the Cytokine Profile of Pathogenic T Cells in Autoimmunity. PLoS ONE 2012, 7, e38708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.H.; Hwang, J.H.; Kim, S.E.; Kim, Y.K.; Park, H.C.; Lee, H.T. Human Galectin-9 on the Porcine Cells Affects the Cytotoxic Activity of M1-Differentiated THP-1 Cells through Inducing a Shift in M2-Differentiated THP-1 Cells. Xenotransplantation 2017, 24, e12305. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Siemann, D.W. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers 2020, 12, 1850. [Google Scholar] [CrossRef]
- Xie, T.; Pan, S.; Zheng, H.; Luo, Z.; Tembo, K.M.; Jamal, M.; Yu, Z.; Yu, Y.; Xia, J.; Yin, Q.; et al. PEG10 as an Oncogene: Expression Regulatory Mechanisms and Role in Tumor Progression. Cancer Cell Int. 2018, 18, 112. [Google Scholar] [CrossRef]
- Gov, E. Co-Expressed Functional Module-Related Genes in Ovarian Cancer Stem Cells Represent Novel Prognostic Biomarkers in Ovarian Cancer. Syst. Biol. Reprod. Med. 2020, 66, 255–266. [Google Scholar] [CrossRef]
- Jiang, Y.; Cao, Y.; Wang, Y.; Li, W.; Liu, X.; Lv, Y.; Li, X.; Mi, J. Cysteine Transporter SLC3A1 Promotes Breast Cancer Tumorigenesis. Theranostics 2017, 7, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Mohan, A.; Raj Rajan, R.; Mohan, G.; Kollenchery Puthenveettil, P.; Maliekal, T.T. Markers and Reporters to Reveal the Hierarchy in Heterogeneous Cancer Stem Cells. Front. Cell Dev. Biol. 2021, 9, 668851. [Google Scholar] [CrossRef]
- Yin, B.W.T.; Lloyd, K.O. Molecular Cloning of the CA125 Ovarian Cancer Antigen. J. Biol. Chem. 2001, 276, 27371–27375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-Associated Fibroblasts: Overview, Progress, Challenges, and Directions. Cancer Gene 2021, 28, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kryczek, I.; Dostál, L.; Lin, H.; Tan, L.; Zhao, L.; Lu, F.; Wei, S.; Maj, T.; Peng, D.; et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell 2016, 165, 1092–1105. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-Associated Fibroblasts Enhance Tumor-Associated Macrophages Enrichment and Suppress NK Cells Function in Colorectal Cancer. Cell Death Dis. 2019, 10, 273. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.J.; Jones, M.G.; Okimoto, R.A.; Nanjo, S.; Chan, M.M.; Yosef, N.; Bivona, T.G.; Weissman, J.S. Single-Cell Lineages Reveal the Rates, Routes, and Drivers of Metastasis in Cancer Xenografts. Science 2021, 371, eabc1944. [Google Scholar] [CrossRef] [PubMed]
- Floderer, M.; Prchal-Murphy, M.; Vizzardelli, C. Dendritic Cell-Secreted Lipocalin2 Induces CD8+ T-Cell Apoptosis, Contributes to T-Cell Priming and Leads to a TH1 Phenotype. PLoS ONE 2014, 9, e101881. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumitani, N.; Ishida, K.; Sawada, K.; Kimura, T.; Kaneda, Y.; Nimura, K. Identification of Malignant Cell Populations Associated with Poor Prognosis in High-Grade Serous Ovarian Cancer Using Single-Cell RNA Sequencing. Cancers 2022, 14, 3580. https://doi.org/10.3390/cancers14153580
Sumitani N, Ishida K, Sawada K, Kimura T, Kaneda Y, Nimura K. Identification of Malignant Cell Populations Associated with Poor Prognosis in High-Grade Serous Ovarian Cancer Using Single-Cell RNA Sequencing. Cancers. 2022; 14(15):3580. https://doi.org/10.3390/cancers14153580
Chicago/Turabian StyleSumitani, Naoki, Kyoso Ishida, Kenjiro Sawada, Tadashi Kimura, Yasufumi Kaneda, and Keisuke Nimura. 2022. "Identification of Malignant Cell Populations Associated with Poor Prognosis in High-Grade Serous Ovarian Cancer Using Single-Cell RNA Sequencing" Cancers 14, no. 15: 3580. https://doi.org/10.3390/cancers14153580
APA StyleSumitani, N., Ishida, K., Sawada, K., Kimura, T., Kaneda, Y., & Nimura, K. (2022). Identification of Malignant Cell Populations Associated with Poor Prognosis in High-Grade Serous Ovarian Cancer Using Single-Cell RNA Sequencing. Cancers, 14(15), 3580. https://doi.org/10.3390/cancers14153580