
SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities


Abstract
:Simple Summary
Abstract
1. Introduction
2. SMARCB1-Deficient Cancers
2.1. Types of SMARCB1-Deficient Cancers
2.2. Initiation of SMARCB1-Deficient Tumors
2.3. Unique Role of SMARCB1 in Synovial Sarcoma
3. Structure of SMARCB1
3.1. Winged Helix Domain
3.2. Tandem Repeat (RPT) Domains
3.3. C-terminal Coiled-Coil Domain (CTD)
3.4. Conservation of SMARCB1
4. SMARCB1 and the BAF Complex
4.1. Three Different BAF Sub-Complexes
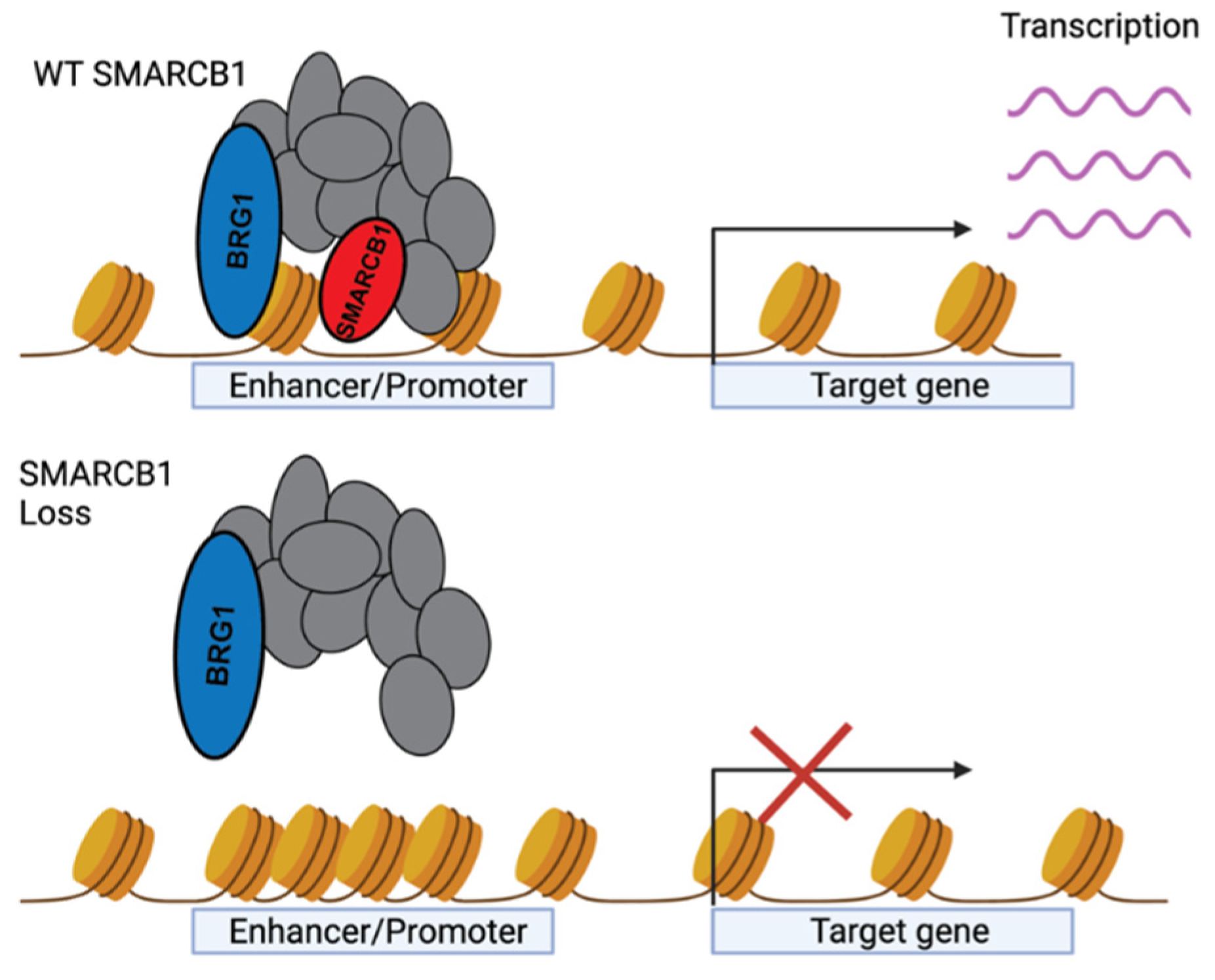
4.2. Transcriptional Regulation by SMARCB1
4.3. SMARCB1 Regulation of Super Enhancers
4.4. SMARCB1-Dependent BAF Complex Stability
5. Molecular Functions of SMARCB1
5.1. SMARCB1 Acting as a Tumor Suppressor via Regulation of p16INK4a
5.2. SMARCB1 Inhibits MYC Target Activation
5.3. Exportin 1 (XPO1)-Mediated Localization of SMARCB1
6. Advances in Molecular Subgrouping of ATRT
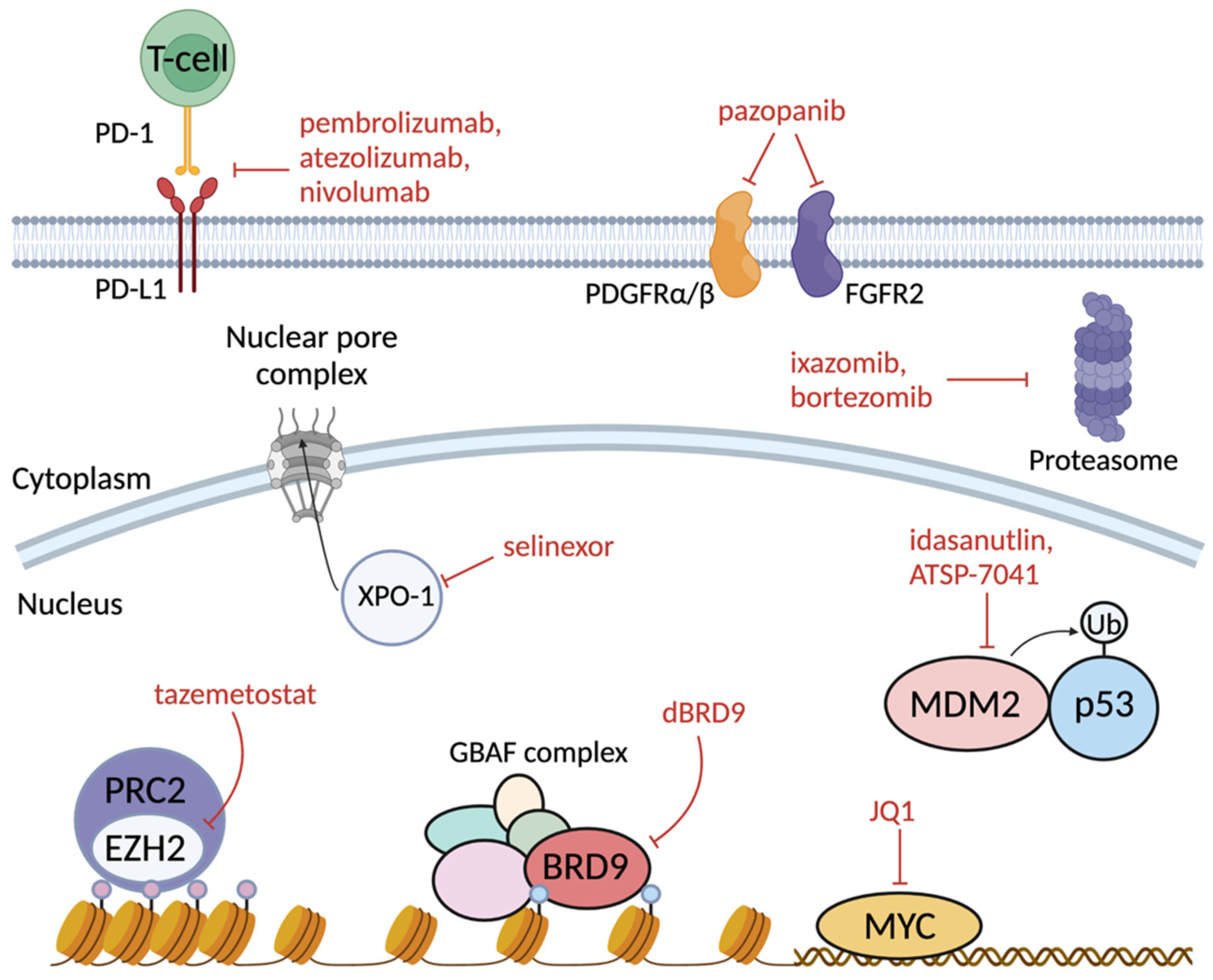
7. Therapeutic Vulnerabilities of SMARCB1-Deficient Cancers
7.1. Opposing Overactive PRC2 Repression by Inhibiting EZH2
7.2. Targeting GBAF Dependency through BRD9 Degradation
7.3. Tyrosine Kinase Inhibition (PDGFRα/β and FGFR2)
7.4. MYC Inhibition
7.5. Immune Checkpoint Inhibition
7.6. High-Throughput Screens to Identify Therapeutic Vulnerabilities
7.6.1. MDM2/4 Inhibition
7.6.2. Proteasomal Inhibition
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Laurent, B.C.; Treitel, M.A.; Carlson, M. The SNF5 protein of Saccharomyces cerevisiae is a glutamine- and proline-rich transcriptional activator that affects expression of a broad spectrum of genes. Mol. Cell Biol. 1990, 10, 5616–5625. [Google Scholar] [CrossRef]
- Neigeborn, L.; Carlson, M. Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genetics 1984, 108, 845–858. [Google Scholar] [CrossRef]
- Kalpana, G.V.; Marmon, S.; Wang, W.; Crabtree, G.R.; Goff, S.P. Binding and Stimulation of HIV-1 Integrase by a Human Homolog of Yeast Transcription Factor SNF5. Science 1994, 266, 2002–2006. [Google Scholar] [CrossRef]
- Versteege, I.; Sévenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef]
- Margol, A.S.; Judkins, A.R. Pathology and diagnosis of SMARCB1-deficient tumors. Cancer Genet. 2014, 207, 358–364. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Biegel, J.A.; Rorke, L.B.; Packer, R.J.; Emanuel, B.S. Monosomy 22 in rhabdoid or atypical tumors of the brain. J. Neurosurg. 1990, 73, 710–714. [Google Scholar] [CrossRef]
- Douglass, E.C.; Valentine, M.; Rowe, S.T.; Parham, D.M.; Wilimas, J.A.; Sanders, J.M.; Houghton, P.J. Malignant rhabdoid tumor: A highly malignant childhood tumor with minimal karyotypic changes. Genes Chromosomes Cancer 1990, 2, 210–216. [Google Scholar] [CrossRef]
- Cheng, J.X.; Tretiakova, M.; Gong, C.; Mandal, S.; Krausz, T.; Taxy, J.B. Renal medullary carcinoma: Rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod. Pathol. 2008, 21, 647–652. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.M.; Folpe, A.L.; Pawel, B.R.; Judkins, A.R.; Biegel, J.A. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod. Pathol. 2013, 26, 385–392. [Google Scholar] [CrossRef]
- AACR Project Genie Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Qin, Y.; Du, F.; Wang, X.; Song, C. Prevalence of SWI/SNF genomic alterations in cancer and association with the response to immune checkpoint inhibitors: A systematic review and meta-analysis. Gene 2022, 834, 146638. [Google Scholar] [CrossRef]
- Biegel, J.A.; Zhou, J.Y.; Rorke, L.B.; Stenstrom, C.; Wainwright, L.M.; Fogelgren, B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999, 59, 74–79. [Google Scholar]
- Hong, A.L.; Tseng, Y.Y.; Wala, J.A.; Kim, W.J.; Kynnap, B.D.; Doshi, M.B.; Kugener, G.; Sandoval, G.J.; Howard, T.P.; Li, J.; et al. Renal medullary carcinomas depend upon SMARCB1 loss and are sensitive to proteasome inhibition. Elife 2019, 8, e44161. [Google Scholar] [CrossRef]
- Johann, P.D.; Hovestadt, V.; Thomas, C.; Jeibmann, A.; Heß, K.; Bens, S.; Oyen, F.; Hawkins, C.; Pierson, C.R.; Aldape, K.; et al. Cribriform neuroepithelial tumor: Molecular characterization of a SMARCB1-deficient non-rhabdoid tumor with favorable long-term outcome. Brain Pathol. 2017, 27, 411–418. [Google Scholar] [CrossRef]
- Cha, Y.J.; Hong, C.K.; Kim, D.S.; Lee, S.K.; Park, H.J.; Kim, S.H. Poorly differentiated chordoma with loss of SMARCB1/INI1 expression in pediatric patients: A report of two cases and review of the literature. Neuropathology 2018, 38, 47–53. [Google Scholar] [CrossRef]
- Kohashi, K.; Oda, Y.; Yamamoto, H.; Tamiya, S.; Matono, H.; Iwamoto, Y.; Taguchi, T.; Tsuneyoshi, M. Reduced expression of SMARCB1/INI1 protein in synovial sarcoma. Mod. Pathol. 2010, 23, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Hulsebos, T.J.; Plomp, A.S.; Wolterman, R.A.; Robanus-Maandag, E.C.; Baas, F.; Wesseling, P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am. J. Hum. Genet. 2007, 80, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonelli, M.; Raso, A.; Mascelli, S.; Gessi, M.; Nozza, P.; Coli, A.; Gardiman, M.P.; Arcella, A.; Massimino, M.; Buttarelli, F.R.; et al. SMARCB1/INI1 Involvement in Pediatric Chordoma: A Mutational and Immunohistochemical Analysis. Am. J. Surg. Pathol. 2017, 41, 56–61. [Google Scholar] [CrossRef]
- Yoshida, M.; Yamashita, D.; Hamakawa, H.; Takahashi, Y.; Yasui, H.; Komatsu, M.; Ohbayashi, C.; Hara, S. SMARCB1-deficient myoepithelial carcinoma of the lung: A case report. Hum. Pathol. Case Rep. 2020, 21, 200414. [Google Scholar] [CrossRef]
- Agaimy, A.; Hartmann, A.; Antonescu, C.R.; Chiosea, S.I.; El-Mofty, S.K.; Geddert, H.; Iro, H.; Lewis, J.S., Jr.; Märkl, B.; Mills, S.E.; et al. SMARCB1 (INI-1)-deficient Sinonasal Carcinoma: A Series of 39 Cases Expanding the Morphologic and Clinicopathologic Spectrum of a Recently Described Entity. Am. J. Surg. Pathol. 2017, 41, 458–471. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Mulvihill, T.S.; Li, L.; Barrott, J.J.; Nelson, M.L.; Wagner, L.; Lock, I.C.; Pozner, A.; Lambert, S.L.; Ozenberger, B.B.; et al. A Role for SMARCB1 in Synovial Sarcomagenesis Reveals that SS18-SSX Induces Canonical BAF Destruction. Cancer Discov. 2021, 11, 2620–2637. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.H.; Tsang, R.K.; Lo, A.W.I.; Chan, S.Y.; Chung, J.C.; Tong, C.C.; Leung, T.W.; Kwong, D.L. SMARCB1 (INI-1)-Deficient Sinonasal Carcinoma: A Systematic Review and Pooled Analysis of Treatment Outcomes. Cancers 2022, 14, 3285. [Google Scholar] [CrossRef] [PubMed]
- Merker, V.L.; Esparza, S.; Smith, M.J.; Stemmer-Rachamimov, A.; Plotkin, S.R. Clinical features of schwannomatosis: A retrospective analysis of 87 patients. Oncologist 2012, 17, 1317–1322. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.W.; Leroux, M.M.; Fleming, M.D.; Orkin, S.H. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell 2002, 2, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.-Y.; Richer, W.; Fréneaux, P.; Chauvin, C.; Lucchesi, C.; Guillemot, D.; Grison, C.; Lequin, D.; Pierron, G.; Masliah-Planchon, J.; et al. The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat. Commun. 2016, 7, 10421. [Google Scholar] [CrossRef] [Green Version]
- Custers, L.; Khabirova, E.; Coorens, T.H.H.; Oliver, T.R.W.; Calandrini, C.; Young, M.D.; Vieira Braga, F.A.; Ellis, P.; Mamanova, L.; Segers, H.; et al. Somatic mutations and single-cell transcriptomes reveal the root of malignant rhabdoid tumours. Nat. Commun. 2021, 12, 1407. [Google Scholar] [CrossRef]
- Nakayama, R.T.; Pulice, J.L.; Valencia, A.M.; McBride, M.J.; McKenzie, Z.M.; Gillespie, M.A.; Ku, W.L.; Teng, M.; Cui, K.; Williams, R.T.; et al. SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat. Genet. 2017, 49, 1613–1623. [Google Scholar] [CrossRef]
- Chasse, M.H.; Johnson, B.K.; Boguslawski, E.A.; Sorensen, K.M.; Rosien, J.E.; Kang, M.H.; Reynolds, C.P.; Heo, L.; Madaj, Z.B.; Beddows, I.; et al. Mithramycin induces promoter reprogramming and differentiation of rhabdoid tumor. EMBO Mol. Med. 2021, 13, e12640. [Google Scholar] [CrossRef]
- Sredni, S.T.; Tomita, T. Rhabdoid tumor predisposition syndrome. Pediatr. Dev. Pathol. 2015, 18, 49–58. [Google Scholar] [CrossRef]
- Del Baldo, G.; Carta, R.; Alessi, I.; Merli, P.; Agolini, E.; Rinelli, M.; Boccuto, L.; Milano, G.M.; Serra, A.; Carai, A.; et al. Rhabdoid Tumor Predisposition Syndrome: From Clinical Suspicion to General Management. Front. Oncol. 2021, 11, 586288. [Google Scholar] [CrossRef]
- Watanabe, I.C.; Billis, A.; Guimarães, M.S.; Alvarenga, M.; de Matos, A.C.; Cardinalli, I.A.; Filippi, R.Z.; de Castro, M.G.; Suzigan, S. Renal medullary carcinoma: Report of seven cases from Brazil. Mod. Pathol. 2007, 20, 914–920. [Google Scholar] [CrossRef] [Green Version]
- de Visscher, S.A.; van Ginkel, R.J.; Wobbes, T.; Veth, R.P.; Ten Heuvel, S.E.; Suurmeijer, A.J.; Hoekstra, H.J. Epithelioid sarcoma: Still an only surgically curable disease. Cancer 2006, 107, 606–612. [Google Scholar] [CrossRef]
- Holland, P.; Merrimen, J.; Pringle, C.; Wood, L.A. Renal medullary carcinoma and its association with sickle cell trait: A case report and literature review. Curr. Oncol. 2020, 27, e53–e56. [Google Scholar] [CrossRef] [Green Version]
- Noujaim, J.; Thway, K.; Bajwa, Z.; Bajwa, A.; Maki, R.G.; Jones, R.L.; Keller, C. Epithelioid Sarcoma: Opportunities for Biology-Driven Targeted Therapy. Front. Oncol. 2015, 5, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Ro, J.Y. The 2020 WHO Classification of Tumors of Soft Tissue: Selected Changes and New Entities. Adv. Anat. Pathol. 2021, 28, 44–58. [Google Scholar] [CrossRef]
- Clark, J.; Rocques, P.J.; Crew, A.J.; Gill, S.; Shipley, J.; Chan, A.M.; Gusterson, B.A.; Cooper, C.S. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat. Genet. 1994, 7, 502–508. [Google Scholar] [CrossRef]
- de Leeuw, B.; Balemans, M.; Olde Weghuis, D.; Geurts van Kessel, A. Identification of two alternative fusion genes, SYT-SSX1 and SYT-SSX2, in t(X;18)(p11.2;q11.2)-positive synovial sarcomas. Hum. Mol. Genet. 1995, 4, 1097–1099. [Google Scholar] [CrossRef]
- Skytting, B.; Nilsson, G.; Brodin, B.; Xie, Y.; Lundeberg, J.; Uhlén, M.; Larsson, O. A novel fusion gene, SYT-SSX4, in synovial sarcoma. J. Natl. Cancer Inst. 1999, 91, 974–975. [Google Scholar] [CrossRef] [Green Version]
- Vlenterie, M.; Hillebrandt-Roeffen, M.H.S.; Flucke, U.E.; Groenen, P.J.T.A.; Tops, B.B.J.; Kamping, E.J.; Pfundt, R.; de Bruijn, D.R.H.; van Kessel, A.H.M.G.; van Krieken, H.J.H.J.M.; et al. Next generation sequencing in synovial sarcoma reveals novel gene mutations. Oncotarget 2015, 6, 34680. [Google Scholar] [CrossRef] [Green Version]
- Kadoch, C.; Crabtree, G.R. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 2013, 153, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; Van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife 2018, 7, e41305. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef]
- Bacci, C.; Sestini, R.; Provenzano, A.; Paganini, I.; Mancini, I.; Porfirio, B.; Vivarelli, R.; Genuardi, M.; Papi, L. Schwannomatosis associated with multiple meningiomas due to a familial SMARCB1 mutation. Neurogenetics 2010, 11, 73–80. [Google Scholar] [CrossRef]
- Christiaans, I.; Kenter, S.B.; Brink, H.C.; van Os, T.A.; Baas, F.; van den Munckhof, P.; Kidd, A.M.; Hulsebos, T.J. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J. Med. Genet. 2011, 48, 93–97. [Google Scholar] [CrossRef]
- Hadfield, K.D.; Newman, W.G.; Bowers, N.L.; Wallace, A.; Bolger, C.; Colley, A.; McCann, E.; Trump, D.; Prescott, T.; Evans, D.G. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J. Med. Genet. 2008, 45, 332–339. [Google Scholar] [CrossRef]
- Smith, M.J.; Walker, J.A.; Shen, Y.; Stemmer-Rachamimov, A.; Gusella, J.F.; Plotkin, S.R. Expression of SMARCB1 (INI1) mutations in familial schwannomatosis. Hum. Mol. Genet. 2012, 21, 5239–5245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.J.; Wallace, A.J.; Bowers, N.L.; Rustad, C.F.; Woods, C.G.; Leschziner, G.D.; Ferner, R.E.; Evans, D.G. Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics 2012, 13, 141–145. [Google Scholar] [CrossRef]
- Teichmann, M.; Dumay-Odelot, H.; Fribourg, S. Structural and functional aspects of winged-helix domains at the core of transcription initiation complexes. Transcription 2012, 3, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.D.; Freund, S.M.; Zinzalla, G.; Bycroft, M. The SWI/SNF Subunit INI1 Contains an N-Terminal Winged Helix DNA Binding Domain that Is a Target for Mutations in Schwannomatosis. Structure 2015, 23, 1344–1349. [Google Scholar] [CrossRef] [Green Version]
- Taylor, I.A.; Treiber, M.K.; Olivi, L.; Smerdon, S.J. The X-ray structure of the DNA-binding domain from the Saccharomyces cerevisiae cell-cycle transcription factor Mbp1 at 2.1 A resolution. J. Mol. Biol. 1997, 272, 891–902. [Google Scholar] [CrossRef]
- He, S.; Wu, Z.; Tian, Y.; Yu, Z.; Yu, J.; Wang, X.; Li, J.; Liu, B.; Xu, Y. Structure of nucleosome-bound human BAF complex. Science 2020, 367, 875–881. [Google Scholar] [CrossRef]
- Mashtalir, N.; Suzuki, H.; Farrell, D.P.; Sankar, A.; Luo, J.; Filipovski, M.; D’Avino, A.R.; St Pierre, R.; Valencia, A.M.; Onikubo, T.; et al. A Structural Model of the Endogenous Human BAF Complex Informs Disease Mechanisms. Cell 2020, 183, 802–817.e824. [Google Scholar] [CrossRef]
- Chen, G.; Zhou, H.; Liu, B.; Wang, Y.; Zhao, J.; Giancotti, F.G.; Long, J. A heterotrimeric SMARCB1–SMARCC2 subcomplex is required for the assembly and tumor suppression function of the BAF chromatin-remodeling complex. Cell Discov. 2020, 6, 66. [Google Scholar] [CrossRef]
- Cheng, S.W.; Davies, K.P.; Yung, E.; Beltran, R.J.; Yu, J.; Kalpana, G.V. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat. Genet. 1999, 22, 102–105. [Google Scholar] [CrossRef]
- Lee, D.; Sohn, H.; Kalpana, G.V.; Choe, J. Interaction of E1 and hSNF5 proteins stimulates replication of human papillomavirus DNA. Nature 1999, 399, 487–491. [Google Scholar] [CrossRef]
- Rozenblatt-Rosen, O.; Rozovskaia, T.; Burakov, D.; Sedkov, Y.; Tillib, S.; Blechman, J.; Nakamura, T.; Croce, C.M.; Mazo, A.; Canaani, E. The C-terminal SET domains of ALL-1 and TRITHORAX interact with the INI1 and SNR1 proteins, components of the SWI/SNF complex. Proc. Natl. Acad. Sci. USA 1998, 95, 4152–4157. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.Y.; Kalpana, G.V.; Goff, S.P.; Schubach, W.H. Epstein-Barr virus nuclear protein 2 (EBNA2) binds to a component of the human SNF-SWI complex, hSNF5/Ini1. J. Virol. 1996, 70, 6020–6028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, M.A.; Taira, T.; Tamai, K.; Iguchi-Ariga, S.M.; Ariga, H. ORC1 interacts with c-Myc to inhibit E-box-dependent transcription by abrogating c-Myc-SNF5/INI1 interaction. Genes Cells 2000, 5, 481–490. [Google Scholar] [CrossRef]
- Craig, E.; Zhang, Z.K.; Davies, K.P.; Kalpana, G.V. A masked NES in INI1/hSNF5 mediates hCRM1-dependent nuclear export: Implications for tumorigenesis. Embo J. 2002, 21, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Valencia, A.M.; Collings, C.K.; Dao, H.T.; St Pierre, R.; Cheng, Y.C.; Huang, J.; Sun, Z.Y.; Seo, H.S.; Mashtalir, N.; Comstock, D.E.; et al. Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling. Cell 2019, 179, 1342–1356.e1323. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Tsurusaki, Y.; Matsumoto, N. Numerous BAF complex genes are mutated in Coffin-Siris syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Tsurusaki, Y.; Okamoto, N.; Ohashi, H.; Mizuno, S.; Matsumoto, N.; Makita, Y.; Fukuda, M.; Isidor, B.; Perrier, J.; Aggarwal, S.; et al. Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin. Genet. 2014, 85, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T.; Okamoto, N. Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Schmitz, U.; Mueller, W.; Weber, M.; Sévenet, N.; Delattre, O.; Deimling, A.V. INI1 mutations in meningiomas at a potential hotspot in exon 9. Br. J. Cancer 2001, 84, 199–201. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Mighell, T.L.; Evans-Dutson, S.; O’Roak, B.J. A Saturation Mutagenesis Approach to Understanding PTEN Lipid Phosphatase Activity and Genotype-Phenotype Relationships. Am. J. Hum. Genet. 2018, 102, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Hernández-García, J.; Diego-Martin, B.; Kuo, P.H.; Jami-Alahmadi, Y.; Vashisht, A.A.; Wohlschlegel, J.; Jacobsen, S.E.; Blázquez, M.A.; Gallego-Bartolomé, J. Comprehensive identification of SWI/SNF complex subunits underpins deep eukaryotic ancestry and reveals new plant components. Commun. Biol. 2022, 5, 549. [Google Scholar] [CrossRef]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zofall, M.; Persinger, J.; Kassabov, S.R.; Bartholomew, B. Chromatin remodeling by ISW2 and SWI/SNF requires DNA translocation inside the nucleosome. Nat. Struct. Mol. Biol. 2006, 13, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Vivas, P.; Dechassa, M.L.; Mooney, A.M.; Poirier, M.G.; Bartholomew, B. The SnAC domain of SWI/SNF is a histone anchor required for remodeling. Mol. Cell Biol. 2013, 33, 360–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef]
- Wang, W.; Côté, J.; Xue, Y.; Zhou, S.; Khavari, P.A.; Biggar, S.R.; Muchardt, C.; Kalpana, G.V.; Goff, S.P.; Yaniv, M.; et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. Embo J. 1996, 15, 5370–5382. [Google Scholar] [CrossRef] [PubMed]
- Lemon, B.; Inouye, C.; King, D.S.; Tjian, R. Selectivity of chromatin-remodelling cofactors for ligand-activated transcription. Nature 2001, 414, 924–928. [Google Scholar] [CrossRef]
- Raab, J.R.; Resnick, S.; Magnuson, T. Genome-Wide Transcriptional Regulation Mediated by Biochemically Distinct SWI/SNF Complexes. PLoS Genet. 2015, 11, e1005748. [Google Scholar] [CrossRef]
- Xue, Y.; Canman, J.C.; Lee, C.S.; Nie, Z.; Yang, D.; Moreno, G.T.; Young, M.K.; Salmon, E.D.; Wang, W. The human SWI/SNF-B chromatin-remodeling complex is related to yeast rsc and localizes at kinetochores of mitotic chromosomes. Proc. Natl. Acad. Sci. USA 2000, 97, 13015–13020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpsoy, A.; Dykhuizen, E.C. Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J. Biol. Chem. 2018, 293, 3892–3903. [Google Scholar] [CrossRef] [Green Version]
- Innis, S.M.; Cabot, B. GBAF, a small BAF sub-complex with big implications: A systematic review. Epigenetics Chromatin 2020, 13, 48. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Troisi, E.C.; Howard, T.P.; Haswell, J.R.; Wolf, B.K.; Hawk, W.H.; Ramos, P.; Oberlick, E.M.; Tzvetkov, E.P.; et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 2019, 10, 1881. [Google Scholar] [CrossRef] [Green Version]
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; St. Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e1220. [Google Scholar] [CrossRef] [Green Version]
- Morrison, E.A.; Sanchez, J.C.; Ronan, J.L.; Farrell, D.P.; Varzavand, K.; Johnson, J.K.; Gu, B.X.; Crabtree, G.R.; Musselman, C.A. DNA binding drives the association of BRG1/hBRM bromodomains with nucleosomes. Nat. Commun. 2017, 8, 16080. [Google Scholar] [CrossRef]
- Tolstorukov, M.Y.; Sansam, C.G.; Lu, P.; Koellhoffer, E.C.; Helming, K.C.; Alver, B.H.; Tillman, E.J.; Evans, J.A.; Wilson, B.G.; Park, P.J.; et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc. Natl. Acad. Sci. USA 2013, 110, 10165–10170. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.M.; Hota, S.K.; He, D.; Thomas, S.; Ho, L.; Pennacchio, L.A.; Bruneau, B.G. Brg1 modulates enhancer activation in mesoderm lineage commitment. Development 2015, 142, 1418–1430. [Google Scholar] [CrossRef] [Green Version]
- Bossen, C.; Murre, C.S.; Chang, A.N.; Mansson, R.; Rodewald, H.-R.; Murre, C. The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat. Immunol. 2015, 16, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Ni, Z.; Abou El Hassan, M.; Xu, Z.; Yu, T.; Bremner, R. The chromatin-remodeling enzyme BRG1 coordinates CIITA induction through many interdependent distal enhancers. Nat. Immunol. 2008, 9, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Chen, Y.; Kim, B.; Wang, H.; Zhao, C.; He, X.; Liu, L.; Liu, W.; Wu, L.M.; Mao, M.; et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 2013, 152, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Alver, B.H.; Kim, K.H.; Lu, P.; Wang, X.; Manchester, H.E.; Wang, W.; Haswell, J.R.; Park, P.J.; Roberts, C.W. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 2017, 8, 14648. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lee, R.S.; Alver, B.H.; Haswell, J.R.; Wang, S.; Mieczkowski, J.; Drier, Y.; Gillespie, S.M.; Archer, T.C.; Wu, J.N.; et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 2017, 49, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Alpsoy, A.; Sood, S.; Dykhuizen, E.C. At the Crossroad of Gene Regulation and Genome Organization: Potential Roles for ATP-Dependent Chromatin Remodelers in the Regulation of CTCF-Mediated 3D Architecture. Biology 2021, 10, 272. [Google Scholar] [CrossRef]
- Alpsoy, A.; Utturkar, S.M.; Carter, B.C.; Dhiman, A.; Torregrosa-Allen, S.E.; Currie, M.P.; Elzey, B.D.; Dykhuizen, E.C. BRD9 Is a Critical Regulator of Androgen Receptor Signaling and Prostate Cancer Progression. Cancer Res. 2021, 81, 820–833. [Google Scholar] [CrossRef]
- Gatchalian, J.; Malik, S.; Ho, J.; Lee, D.S.; Kelso, T.W.R.; Shokhirev, M.N.; Dixon, J.R.; Hargreaves, D.C. A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun. 2018, 9, 5139. [Google Scholar] [CrossRef] [Green Version]
- Inoue, D.; Chew, G.L.; Liu, B.; Michel, B.C.; Pangallo, J.; D’Avino, A.R.; Hitchman, T.; North, K.; Lee, S.C.; Bitner, L.; et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 2019, 574, 432–436. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Gates, L.A.; Foulds, C.E.; O’Malley, B.W. Histone Marks in the ‘Driver’s Seat’: Functional Roles in Steering the Transcription Cycle. Trends Biochem. Sci. 2017, 42, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Beacon, T.H.; Delcuve, G.P.; López, C.; Nardocci, G.; Kovalchuk, I.; van Wijnen, A.J.; Davie, J.R. The dynamic broad epigenetic (H3K4me3, H3K27ac) domain as a mark of essential genes. Clin. Epigenetics 2021, 13, 138. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Wysocka, J. Modification of Enhancer Chromatin: What, How, and Why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Kadoch, C.; Williams, R.T.; Calarco, J.P.; Miller, E.L.; Weber, C.M.; Braun, S.M.G.; Pulice, J.L.; Chory, E.J.; Crabtree, G.R. Dynamics of BAF–Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet. 2017, 49, 213–222. [Google Scholar] [CrossRef]
- Zhang, W.; Chronis, C.; Chen, X.; Zhang, H.; Spalinskas, R.; Pardo, M.; Chen, L.; Wu, G.; Zhu, Z.; Yu, Y.; et al. The BAF and PRC2 Complex Subunits Dpf2 and Eed Antagonistically Converge on Tbx3 to Control ESC Differentiation. Cell Stem. Cell 2019, 24, 138–152.e138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kia, S.K.; Gorski, M.M.; Giannakopoulos, S.; Verrijzer, C.P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell Biol. 2008, 28, 3457–3464. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Sansam, C.G.; Thom, C.S.; Metzger, D.; Evans, J.A.; Nguyen, P.T.; Roberts, C.W. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009, 69, 8094–8101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkek, S.; Johann, P.D.; Finetti, M.A.; Drosos, Y.; Chou, H.C.; Zapatka, M.; Sturm, D.; Jones, D.T.W.; Korshunov, A.; Rhyzova, M.; et al. Comprehensive Analysis of Chromatin States in Atypical Teratoid/Rhabdoid Tumor Identifies Diverging Roles for SWI/SNF and Polycomb in Gene Regulation. Cancer Cell 2019, 35, 95–110.e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M. Transcriptional enhancers in animal development and evolution. Curr. Biol. 2010, 20, R754–R763. [Google Scholar] [CrossRef] [Green Version]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Langer, L.F.; Ward, J.M.; Archer, T.K. Tumor suppressor SMARCB1 suppresses super-enhancers to govern hESC lineage determination. ELife 2019, 8, e45672. [Google Scholar] [CrossRef]
- Sen, P.; Luo, J.; Hada, A.; Hailu, S.G.; Dechassa, M.L.; Persinger, J.; Brahma, S.; Paul, S.; Ranish, J.; Bartholomew, B. Loss of Snf5 Induces Formation of an Aberrant SWI/SNF Complex. Cell Rep. 2017, 18, 2135–2147. [Google Scholar] [CrossRef]
- Wei, D.; Goldfarb, D.; Song, S.; Cannon, C.; Yan, F.; Sakellariou-Thompson, D.; Emanuele, M.; Major, M.B.; Weissman, B.E.; Kuwahara, Y. SNF5/INI1 Deficiency Redefines Chromatin Remodeling Complex Composition during Tumor Development. Mol. Cancer Res. 2014, 12, 1574–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doan, D.N.; Veal, T.M.; Yan, Z.; Wang, W.; Jones, S.N.; Imbalzano, A.N. Loss of the INI1 tumor suppressor does not impair the expression of multiple BRG1-dependent genes or the assembly of SWI/SNF enzymes. Oncogene 2004, 23, 3462–3473. [Google Scholar] [CrossRef] [Green Version]
- Jamshidi, F.; Bashashati, A.; Shumansky, K.; Dickson, B.; Gokgoz, N.; Wunder, J.S.; Andrulis, I.L.; Lazar, A.J.; Shah, S.P.; Huntsman, D.G.; et al. The genomic landscape of epithelioid sarcoma cell lines and tumours. J. Pathol. 2016, 238, 63–73. [Google Scholar] [CrossRef]
- Betz, B.L.; Strobeck, M.W.; Reisman, D.N.; Knudsen, E.S.; Weissman, B.E. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene 2002, 21, 5193–5203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagosa, C.; Simonetti, S.; López-Vicente, L.; Mazo, A.; Lleonart, M.E.; Castellvi, J.; Ramon y Cajal, S. p16Ink4a overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 2011, 30, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGregori, J.; Leone, G.; Miron, A.; Jakoi, L.; Nevins, J.R. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 7245–7250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venneti, S.; Le, P.; Martinez, D.; Eaton, K.W.; Shyam, N.; Jordan-Sciutto, K.L.; Pawel, B.; Biegel, J.A.; Judkins, A.R. p16INK4A and p14ARF tumor suppressor pathways are deregulated in malignant rhabdoid tumors. J. Neuropathol. Exp. Neurol. 2011, 70, 596–609. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadd, S.; Sredni, S.T.; Huang, C.C.; Perlman, E.J. Rhabdoid tumor: Gene expression clues to pathogenesis and potential therapeutic targets. Lab. Investig. 2010, 90, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Werneck, M.B.; Wilson, B.G.; Kim, H.J.; Kluk, M.J.; Thom, C.S.; Wischhusen, J.W.; Evans, J.A.; Jesneck, J.L.; Nguyen, P.; et al. TCR-dependent transformation of mature memory phenotype T cells in mice. J. Clin. Investig. 2011, 121, 3834–3845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Carugo, A.; Tepper, J.; Robinson, F.S.; Li, L.; Svelto, M.; Nezi, L.; Corti, D.; Minelli, R.; Pettazzoni, P.; et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 2017, 542, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Stojanova, A.; Tu, W.B.; Ponzielli, R.; Kotlyar, M.; Chan, P.K.; Boutros, P.C.; Khosravi, F.; Jurisica, I.; Raught, B.; Penn, L.Z. MYC interaction with the tumor suppressive SWI/SNF complex member INI1 regulates transcription and cellular transformation. Cell Cycle 2016, 15, 1693–1705. [Google Scholar] [CrossRef] [PubMed]
- Weissmiller, A.M.; Wang, J.; Lorey, S.L.; Howard, G.C.; Martinez, E.; Liu, Q.; Tansey, W.P. Inhibition of MYC by the SMARCB1 tumor suppressor. Nat. Commun. 2019, 10, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammak, S.; Allen, M.D.; Hamdani, N.; Bycroft, M.; Zinzalla, G. The structure of INI1/hSNF5 RPT1 and its interactions with the c-MYC:MAX heterodimer provide insights into the interplay between MYC and the SWI/SNF chromatin remodeling complex. Febs J. 2018, 285, 4165–4180. [Google Scholar] [CrossRef]
- Judkins, A.R.; Mauger, J.; Ht, A.; Rorke, L.B.; Biegel, J.A. Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am. J. Surg. Pathol. 2004, 28, 644–650. [Google Scholar] [CrossRef]
- Ho, B.; Johann, P.D.; Grabovska, Y.; De Dieu Andrianteranagna, M.J.; Yao, F.; Frühwald, M.; Hasselblatt, M.; Bourdeaut, F.; Williamson, D.; Huang, A.; et al. Molecular subgrouping of atypical teratoid/rhabdoid tumors-a reinvestigation and current consensus. Neurooncology 2020, 22, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Pathak, R.; Zin, F.; Thomas, C.; Bens, S.; Gayden, T.; Karamchandani, J.; Dudley, R.W.; Nemes, K.; Johann, P.D.; Oyen, F.; et al. Inhibition of nuclear export restores nuclear localization and residual tumor suppressor function of truncated SMARCB1/INI1 protein in a molecular subset of atypical teratoid/rhabdoid tumors. Acta Neuropathol. 2021, 142, 361–374. [Google Scholar] [CrossRef]
- Hilden, J.M.; Meerbaum, S.; Burger, P.; Finlay, J.; Janss, A.; Scheithauer, B.W.; Walter, A.W.; Rorke, L.B.; Biegel, J.A. Central Nervous System Atypical Teratoid/Rhabdoid Tumor: Results of Therapy in Children Enrolled in a Registry. J. Clin. Oncol. 2004, 22, 2877–2884. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.M.T.; Hsu, C.Y.; Wong, T.T.; Ting, L.T.; Chiang, H. Atypical teratoid/rhabdoid tumor of the central nervous system: A comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol. 2000, 99, 482–488. [Google Scholar] [CrossRef]
- Packer, R.J.; Biegel, J.A.; Blaney, S.; Finlay, J.; Geyer, J.R.; Heideman, R.; Hilden, J.; Janss, A.J.; Kun, L.; Vezina, G.; et al. Atypical Teratoid/Rhabdoid Tumor of the Central Nervous System: Report on Workshop. J. Pediatric Hematol. Oncol. 2002, 24, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Birks, D.K.; Donson, A.M.; Patel, P.R.; Dunham, C.; Muscat, A.; Algar, E.M.; Ashley, D.M.; Kleinschmidt-Demasters, B.K.; Vibhakar, R.; Handler, M.H.; et al. High expression of BMP pathway genes distinguishes a subset of atypical teratoid/rhabdoid tumors associated with shorter survival. Neurooncology 2011, 13, 1296–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.W.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchia, J.; Golbourn, B.; Feng, S.; Ho, K.C.; Sin-Chan, P.; Vasiljevic, A.; Norman, J.D.; Guilhamon, P.; Garzia, L.; Agamez, N.R.; et al. Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell 2016, 30, 891–908. [Google Scholar] [CrossRef] [Green Version]
- Torchia, J.; Picard, D.; Lafay-Cousin, L.; Hawkins, C.E.; Kim, S.K.; Letourneau, L.; Ra, Y.S.; Ho, K.C.; Chan, T.S.; Sin-Chan, P.; et al. Molecular subgroups of atypical teratoid rhabdoid tumours in children: An integrated genomic and clinicopathological analysis. Lancet Oncol. 2015, 16, 569–582. [Google Scholar] [CrossRef]
- Upadhyaya, S.A.; Robinson, G.W.; Onar-Thomas, A.; Orr, B.A.; Johann, P.; Wu, G.; Billups, C.A.; Tatevossian, R.G.; Dhanda, S.K.; Srinivasan, A.; et al. Relevance of Molecular Groups in Children with Newly Diagnosed Atypical Teratoid Rhabdoid Tumor: Results from Prospective St. Jude Multi-institutional Trials. Clin. Cancer Res. 2021, 27, 2879–2889. [Google Scholar] [CrossRef]
- Hasselblatt, M.; Thomas, C.; Nemes, K.; Monoranu, C.-M.; Riemenschneider, M.J.; Koch, A.; Sumerauer, D.; Hauser, P.; Paulus, W.; Johann, P.D.; et al. Tyrosinase immunohistochemistry can be employed for the diagnosis of atypical teratoid/rhabdoid tumours of the tyrosinase subgroup (ATRT-TYR). Neuropathol. Appl. Neurobiol. 2020, 46, 186–189. [Google Scholar] [CrossRef]
- Ingham, P.W.; Nakano, Y.; Seger, C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet. 2011, 12, 393–406. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Holdhof, D.; Johann, P.D.; Spohn, M.; Bockmayr, M.; Safaei, S.; Joshi, P.; Masliah-Planchon, J.; Ho, B.; Andrianteranagna, M.; Bourdeaut, F.; et al. Atypical teratoid/rhabdoid tumors (ATRTs) with SMARCA4 mutation are molecularly distinct from SMARCB1-deficient cases. Acta Neuropathol. 2021, 141, 291–301. [Google Scholar] [CrossRef]
- Zhang, C.; Li, H. Molecular targeted therapies for pediatric atypical teratoid/rhabdoid tumors. Pediatric Investig. 2022, 6, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.; Schöffski, P.; Jones, R.L.; Agulnik, M.; Cote, G.M.; Villalobos, V.M.; Attia, S.; Chugh, R.; Chen, T.W.; Jahan, T.; et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: An international, open-label, phase 2 basket study. Lancet Oncol. 2020, 21, 1423–1432. [Google Scholar] [CrossRef]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Porter Scott, M.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [Green Version]
- Hoy, S.M. Tazemetostat: First Approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Straining, R.; Eighmy, W. Tazemetostat: EZH2 Inhibitor. J. Adv. Pract. Oncol. 2022, 13, 158–163. [Google Scholar] [CrossRef]
- Krämer, K.F.; Moreno, N.; Frühwald, M.C.; Kerl, K. BRD9 Inhibition, Alone or in Combination with Cytostatic Compounds as a Therapeutic Approach in Rhabdoid Tumors. Int. J. Mol. Sci. 2017, 18, 1537. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.P.; Todd, J.R.; Finetti, M.A.; McCarthy, F.; Broncel, M.; Vyse, S.; Luczynski, M.T.; Crosier, S.; Ryall, K.A.; Holmes, K.; et al. Dual Targeting of PDGFRα and FGFR1 Displays Synergistic Efficacy in Malignant Rhabdoid Tumors. Cell Rep. 2016, 17, 1265–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Lun, X.; Jayanthan, A.; Obaid, H.; Ruan, Y.; Strother, D.; Chi, S.N.; Smith, A.; Forsyth, P.; Narendran, A. Profiling pathway-specific novel therapeutics in preclinical assessment for central nervous system atypical teratoid rhabdoid tumors (CNS ATRT): Favorable activity of targeting EGFR-ErbB2 signaling with lapatinib. Mol. Oncol. 2013, 7, 497–512. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [Green Version]
- Alimova, I.; Pierce, A.; Danis, E.; Donson, A.; Birks, D.K.; Griesinger, A.; Foreman, N.K.; Santi, M.; Soucek, L.; Venkataraman, S.; et al. Inhibition of MYC attenuates tumor cell self-renewal and promotes senescence in SMARCB1-deficient Group 2 atypical teratoid rhabdoid tumors to suppress tumor growth in vivo. Int. J. Cancer 2019, 144, 1983–1995. [Google Scholar] [CrossRef]
- Leruste, A.; Tosello, J.; Ramos, R.N.; Tauziède-Espariat, A.; Brohard, S.; Han, Z.Y.; Beccaria, K.; Andrianteranagna, M.; Caudana, P.; Nikolic, J.; et al. Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer Cell 2019, 36, 597–612.e598. [Google Scholar] [CrossRef]
- Chun, H.-J.E.; Johann, P.D.; Milne, K.; Zapatka, M.; Buellesbach, A.; Ishaque, N.; Iskar, M.; Erkek, S.; Wei, L.; Tessier-Cloutier, B.; et al. Identification and Analyses of Extra-Cranial and Cranial Rhabdoid Tumor Molecular Subgroups Reveal Tumors with Cytotoxic T Cell Infiltration. Cell Rep. 2019, 29, 2338–2354.e2337. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Malouf, G.G.; Su, X.; Yao, H.; Tripathi, D.N.; Soeung, M.; Gao, J.; Rao, P.; Coarfa, C.; Creighton, C.J.; et al. Comprehensive Molecular Characterization Identifies Distinct Genomic and Immune Hallmarks of Renal Medullary Carcinoma. Cancer Cell 2020, 37, 720–734.e713. [Google Scholar] [CrossRef]
- Forrest, S.J.; Al-Ibraheemi, A.; Doan, D.; Ward, A.; Clinton, C.M.; Putra, J.; Pinches, R.S.; Kadoch, C.; Chi, S.N.; DuBois, S.G.; et al. Genomic and Immunologic Characterization of INI1-Deficient Pediatric Cancers. Clin. Cancer Res. 2020, 26, 2882–2890. [Google Scholar] [CrossRef] [Green Version]
- Ngo, C.; Postel-Vinay, S. Immunotherapy for SMARCB1-Deficient Sarcomas: Current Evidence and Future Developments. Biomedicines 2022, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.P.; Arnoff, T.E.; Song, M.R.; Giacomelli, A.O.; Wang, X.F.; Hong, A.L.; Dharia, N.V.; Wang, S.; Vazquez, F.; Pham, M.T.; et al. MDM2 and MDM4 Are Therapeutic Vulnerabilities in Malignant Rhabdoid Tumors. Cancer Res. 2019, 79, 2404–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carugo, A.; Minelli, R.; Sapio, L.; Soeung, M.; Carbone, F.; Robinson, F.S.; Tepper, J.; Chen, Z.; Lovisa, S.; Svelto, M.; et al. p53 Is a Master Regulator of Proteostasis in SMARCB1-Deficient Malignant Rhabdoid Tumors. Cancer Cell 2019, 35, 204–220.e209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, A.; Soane, C.; Pierce, A.; Sanford, B.; Jones, K.L.; Crespo, M.; Zahedi, S.; Vibhakar, R.; Mulcahy Levy, J.M. Proteasome inhibition as a therapeutic approach in atypical teratoid/rhabdoid tumors. Neurooncol. Adv. 2020, 2, vdaa051. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KERRYPNX | ATRT-MYC | ATRT-SHH | ATRT-TYR | Reference(s) |
|---|---|---|---|---|
| Median age at diagnosis (years) | 3.4 | 1.4 | 1.5 | [136] |
| % with RTPS | 0% | ~36% | ~20% | [136] |
| % Metastatic | ~30% | ~46% | ~10% | [136] |
| Predominant CNV at SMARCB1 locus | Focal loss (50%) | Focal loss (50%) | Focal (5%) | [133,136] |
| Broad loss (7%) | Broad loss (7%) | Broad loss (62%) | ||
| Small loss (29%) | Small loss (29%) | Small loss (20%) | ||
| None (14%) | None (14%) | None (24%) | ||
| Site of tumor | Infratentorial (22%) | Infratentorial (30%) | Infratentorial (80%) | [133,136] |
| Supratentorial (64%) | Supratentorial (70%) | Supratentorial (20%) | ||
| Spine (14%) | ||||
| Molecular characterization | Overexpression of the MYC oncogene and HOX cluster | Overexpression of sonic hedgehog and notch members | Overexpression of tyrinosinase and melanosomal gene | [127,133] |
| Sex | Male (54%) | Male (47%) | Male (62%) | [136] |
| Female (46%) | Female (53%) | Female (38%) | ||
| 5-year Overall Survival | 16.7 ± 10.8% | 15 ± 9.8% | 58.8 ± 11.9% | [136] |
| Methylation Status | Hypomethylated | Hypermethylated | Hypermethylated | [133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooper, G.W.; Hong, A.L. SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers 2022, 14, 3645. https://doi.org/10.3390/cancers14153645
Cooper GW, Hong AL. SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers. 2022; 14(15):3645. https://doi.org/10.3390/cancers14153645
Chicago/Turabian StyleCooper, Garrett W., and Andrew L. Hong. 2022. "SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities" Cancers 14, no. 15: 3645. https://doi.org/10.3390/cancers14153645
APA StyleCooper, G. W., & Hong, A. L. (2022). SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers, 14(15), 3645. https://doi.org/10.3390/cancers14153645