
Clinical Outcomes of Patients with Recurrent Microsatellite-Stable Endometrial Cancer in Early-Phase Immunotherapy Clinical Trials

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Population
2.2. Clinical Data Extraction
2.3. Statistical Analysis
3. Results
3.1. Toxicity
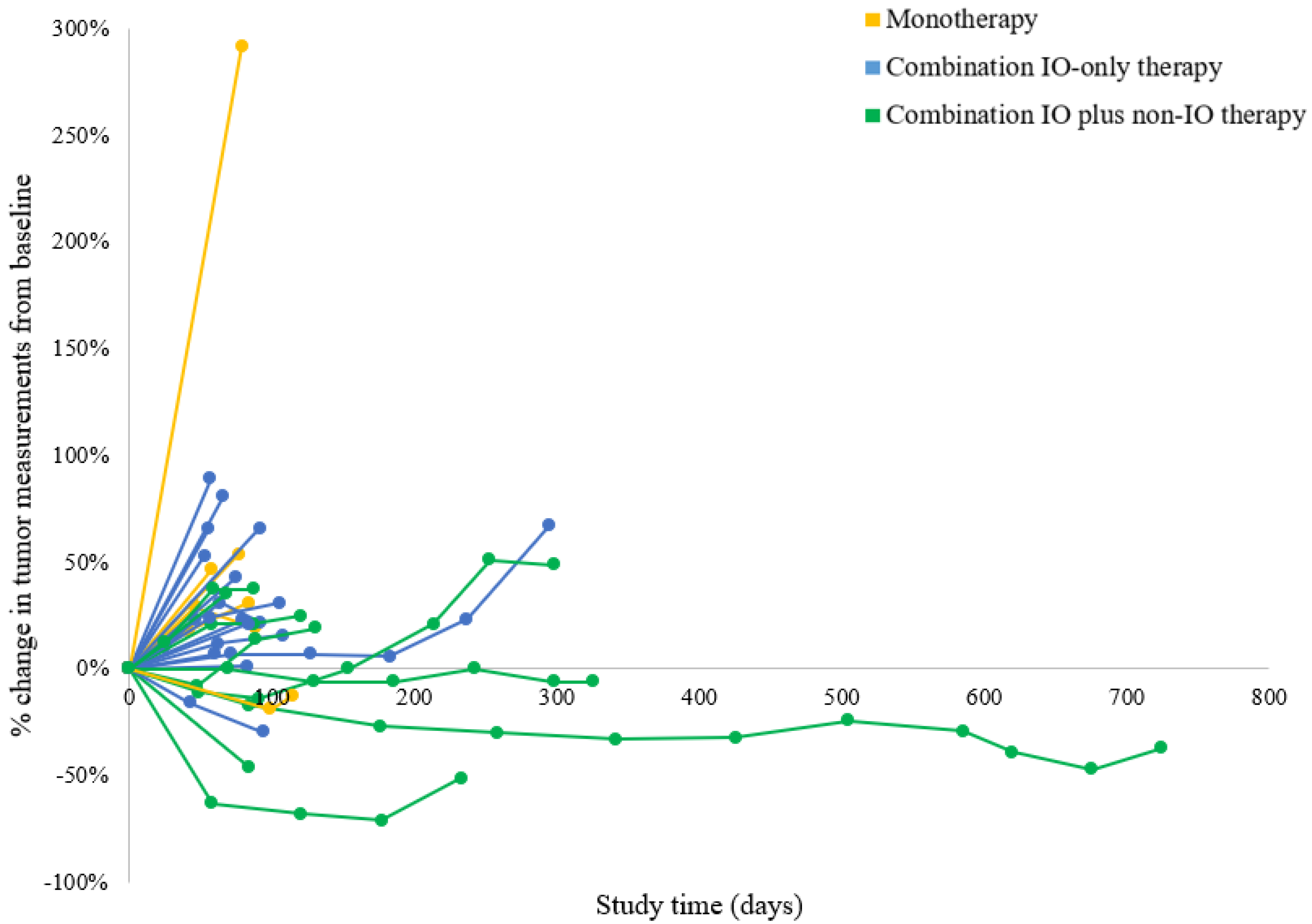
3.2. Clinical Efficacy and Survival
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dizon, D.S. Treatment options for advanced endometrial carcinoma. Gynecol. Oncol. 2010, 117, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, T.H.; Monk, B.J. Systemic therapy for recurrent endometrial cancer: A review of North American trials. Expert Rev. Anticancer Ther. 2009, 9, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Oaknin, A.; Tinker, A.V.; Gilbert, L.; Samouëlian, V.; Mathews, C.; Brown, J.; Barretina-Ginesta, M.P.; Moreno, V.; Gravina, A.; Abdeddaim, C.; et al. Clinical Activity and Safety of the Anti-Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients with Recurrent or Advanced Mismatch Repair-Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA Oncol. 2020, 6, 1766–1772. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Luo, W.; Liu, J.F.; Gulhan, D.C.; Krasner, C.; Ishizuka, J.J.; Gockley, A.A.; Buss, M.; Growdon, W.B.; Crowe, H.; et al. Phase II Study of Avelumab in Patients with Mismatch Repair Deficient and Mismatch Repair Proficient Recurrent/Persistent Endometrial Cancer. J. Clin. Oncol. 2019, 37, 2786–2794. [Google Scholar] [CrossRef] [PubMed]
- Azad, N.S.; Gray, R.J.; Overman, M.J.; Schoenfeld, J.D.; Mitchell, E.P.; Zwiebel, J.A.; Sharon, E.; Streicher, H.; Li, S.; McShane, L.M.; et al. Nivolumab Is Effective in Mismatch Repair-Deficient Noncolorectal Cancers: Results from Arm Z1D-A Subprotocol of the NCI-MATCH (EAY131) Study. J. Clin. Oncol. 2020, 38, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Antill, Y.C.; Kok, P.S.; Robledo, K.; Barnes, E.; Friedlander, M.; Baron-Hay, S.E.; Shannon, C.M.; Coward, J.; Beale, P.J.; Goss, G.; et al. Activity of durvalumab in advanced endometrial cancer (AEC) according to mismatch repair (MMR) status: The phase II PHAEDRA trial (ANZGOG1601). J. Clin. Oncol. 2019, 37, 5501. [Google Scholar] [CrossRef]
- Tamura, K.; Hasegawa, K.; Katsumata, N.; Matsumoto, K.; Mukai, H.; Takahashi, S.; Nomura, H.; Minami, H. Efficacy and safety of nivolumab in Japanese patients with uterine cervical cancer, uterine corpus cancer, or soft tissue sarcoma: Multicenter, open-label phase 2 trial. Cancer Sci. 2019, 110, 2894–2904. [Google Scholar] [CrossRef]
- Ott, P.A.; Bang, Y.J.; Berton-Rigaud, D.; Elez, E.; Pishvaian, M.J.; Rugo, H.S.; Puzanov, I.; Mehnert, J.M.; Aung, K.L.; Lopez, J.; et al. Safety and Antitumor Activity of Pembrolizumab in Advanced Programmed Death Ligand 1-Positive Endometrial Cancer: Results from the KEYNOTE-028 Study. J. Clin. Oncol. 2017, 35, 2535–2541. [Google Scholar] [CrossRef]
- Kunitomi, H.; Banno, K.; Yanokura, M.; Takeda, T.; Iijima, M.; Nakamura, K.; Iida, M.; Adachi, M.; Watanabe, K.; Matoba, Y.; et al. New use of microsatellite instability analysis in endometrial cancer. Oncol. Lett. 2017, 14, 3297–3301. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N.; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbé, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Zehir, A.; Berger, M.F.; Seshan, V.E.; Chan, T.A.; Morris, L.G.T. Response Rates to Anti-PD-1 Immunotherapy in Microsatellite-Stable Solid Tumors with 10 or More Mutations per Megabase. JAMA Oncol. 2021, 7, 739–743. [Google Scholar] [CrossRef]
- Shia, J.; Black, D.; Hummer, A.J.; Boyd, J.; Soslow, R.A. Routinely assessed morphological features correlate with microsatellite instability status in endometrial cancer. Hum. Pathol. 2008, 39, 116–125. [Google Scholar] [CrossRef]
- Nowicki, T.S.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. 2018, 24, 47–53. [Google Scholar] [CrossRef]
- Horton, B.L.; Williams, J.B.; Cabanov, A.; Spranger, S.; Gajewski, T.F. Intratumoral CD8(+) T-cell Apoptosis Is a Major Component of T-cell Dysfunction and Impedes Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Verma, V.; Shrimali, R.K.; Ahmad, S.; Dai, W.; Wang, H.; Lu, S.; Nandre, R.; Gaur, P.; Lopez, J.; Sade-Feldman, M.; et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat. Immunol. 2019, 20, 1231–1243. [Google Scholar] [CrossRef]
- Tang, T.; Huang, X.; Zhang, G.; Hong, Z.; Bai, X.; Liang, T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 72. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musacchio, L.; Caruso, G.; Pisano, C.; Cecere, S.C.; Di Napoli, M.; Attademo, L.; Tambaro, R.; Russo, D.; Califano, D.; Palaia, I.; et al. PARP Inhibitors in Endometrial Cancer: Current Status and Perspectives. Cancer Manag. Res. 2020, 12, 6123–6135. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Yélamos, J.; Moreno-Lama, L.; Jimeno, J.; Ali, S.O. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers 2020, 12, 392. [Google Scholar] [CrossRef] [Green Version]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J. Hematol. Oncol. 2019, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Maenhout, S.K.; Thielemans, K.; Aerts, J.L. Location, location, location: Functional and phenotypic heterogeneity between tumor-infiltrating and non-infiltrating myeloid-derived suppressor cells. Oncoimmunology 2014, 3, e956579. [Google Scholar] [CrossRef]
- Makker, V.; Taylor, M.H.; Aghajanian, C.; Oaknin, A.; Mier, J.; Cohn, A.L.; Romeo, M.; Bratos, R.; Brose, M.S.; DiSimone, C.; et al. Lenvatinib Plus Pembrolizumab in Patients with Advanced Endometrial Cancer. J. Clin. Oncol. 2020, 38, 2981–2992. [Google Scholar] [CrossRef]
- Awad, E.; Paladugu, R.; Jones, N.; Pierce, J.Y.; Scalici, J.; Hamilton, C.A.; Darcy, K.M.; Maxwell, G.L.; Rocconi, R.P. Minority participation in phase 1 gynecologic oncology clinical trials: Three decades of inequity. Gynecol. Oncol. 2020, 157, 729–732. [Google Scholar] [CrossRef]
- Lu, K.H.; Broaddus, R.R. Endometrial Cancer. N. Engl. J. Med. 2020, 383, 2053–2064. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Overall (n = 35) | Monotherapy (n = 8) | Combination | p-Value | ||
|---|---|---|---|---|---|
| IO-Only Therapy (n = 17) | IO Plus Non-IO Therapy (n = 10) | ||||
| Age (median, range), years | 64 (37–73) | 65 (54–73) | 64 (37–71) | 56 (44–68) | 0.2111 k |
| Race/ethnicity | 0.309 f | ||||
| Non-Hispanic White | 20 (57.1%) | 7 (87.5%) | 8 (47.1%) | 5 (50.0%) | |
| Black | 6 (17.1%) | 0 (0.0%) | 5 (29.4%) | 1 (10.0%) | |
| Hispanic | 5 (14.3%) | 0 (0.0%) | 2 (11.8%) | 3 (30.0%) | |
| Asian | 4 (11.4%) | 1 (12.5%) | 2 (11.8%) | 1 (10.0%) | |
| ECOG performance status | >0.999 f | ||||
| 0 | 1 (2.9%) | 0 (0.0%) | 1 (5.9%) | 0 (0.0%) | |
| 1 | 34 (97.1%) | 8 (100%) | 16 (94.1%) | 10 (100%) | |
| MSI status | 0.229 f | ||||
| MSS | 34 (97.1%) | 7 (87.5%) | 17 (100%) | 10 (100%) | |
| MSI unknown | 1 (2.9%) | 1 (12.5%) | 0 (0.0%) | 0 (0.0%) | |
| Histologic type | 0.357 f | ||||
| Endometrioid | 15 (42.9%) | 2 (25.0%) | 6 (35.3%) | 7 (70.0%) | |
| Serous | 8 (22.9%) | 3 (37.5%) | 4 (23.5%) | 1 (10.0%) | |
| Clear cell | 5 (14.3%) | 1 (12.5%) | 4 (23.5%) | 0 (0.0%) | |
| Carcinosarcoma | 4 (11.4%) | 1 (12.5%) | 1 (5.9%) | 2 (20.0%) | |
| Mixed | 3 (8.6%) | 1 (12.5%) | 2 (11.8%) | 0 (0.0%) | |
| Tumor grade | 0.832 f | ||||
| 2 | 5 (14.3%) | 1 (12.5%) | 2 (11.8%) | 2 (20.0%) | |
| 3 | 30 (85.7%) | 7 (87.5%) | 15 (88.2%) | 8 (80.0%) | |
| Prior treatment | 0.1428 k | ||||
| Lines of systemic therapy (median, range) | 3 (1–10) | 3 (2–7) | 4 (1–10) | 2 (1–5) | |
| Number of immune checkpoint inhibitors | <0.001 f | ||||
| None | 1 (2.9%) | 0 (0.0%) | 0 (0.0%) | 1 (10.0%) | |
| 1 | 19 (54.3%) | 8 (100%) | 2 (11.8%) | 9 (90.0%) | |
| 2 | 12 (34.3%) | 0 (0.0%) | 12 (70.6%) | 0 (0.0%) | |
| 3 | 3 (8.6%) | 0 (0.0%) | 3 (17.6%) | 0 (0.0%) | |
| Response Category | Monotherapy (n = 6) | Combination Therapy | p-Value | |
|---|---|---|---|---|
| IO Only (n = 16) | IO Plus Non-IO (n = 10) | |||
| PR | 0 (0%) | 0 (0%) | 1 (10.0%) | 0.337 f |
| CR | 0 (0%) | 0 (0%) | 0 (0.0%) | |
| SD | 1 (16.7%) | 3 (18.8%) | 4 (40.0%) | |
| PD | 5 (83.3%) | 13 (81.2%) | 5 (50.0%) | |
| ORR | 0 (0%) | 0 (0%) | 1 (10.0%) | 0.500 f |
| CBR | 1 (16.7%) | 3 (18.8%) | 5 (50.0%) | 0.197 f |
| Regimen | Group | Histo | Duration of Clinical Benefit (Months) | OS (Months) | Mutations |
|---|---|---|---|---|---|
| -Anti-PD-L1 inhibitor -PARP inhibitor -VEGF inhibitor | IO plus non-IO | E | 21.0 | 24 | Inactivating: PTEN (c.306del) Activating: PIK3CA (c.328_330del and c.1616C > G) |
| -Anti-PD-L1 inhibitor -PARP inhibitor | IO plus non-IO | S | 8.4 | 12 | Inactivating: BRCA1 (c.1513) and TP53 (p.R273H) Unknown functional significance: RAD54L, ESR1, SYK, MAP3K1, and MLL |
| -Anti-PD-L1 inhibitor -Tyrosine kinase inhibitor | IO plus non-IO | E | 8.1 | 18 | Inactivating: PTEN (c.697C > T) and FBXW7 (c.2065C > T) Activating: KRAS (c.35G > C) |
| -Anti-PD-L1 inhibitor -OX40 agonist | Combo IO | M | 7.4 | 14 | Inactivating: FBXW7 (c.1514G > A) Benign: TP53 |
| -Anti-PD-L1 inhibitor -PARP inhibitor | IO plus non-IO | CS | 5.8 | 12 | Inactivating: PTEN (c.955_958del), BRCA1 (c.3013G > T), and PIK3R1 (c.1393_1401del) Unknown functional significance: AXL and NF1 |
| -Anti-PD-L1 inhibitor -Tyrosine kinase inhibitor | IO plus non-IO | E | 2.7 | 3.4 | Inactivating: PTEN (c.388C > T and c.295G > T p.E99), TP53 (c.578A > G), and PIK3R1 (c.1699A > G) Unknown functional significance: CTNNB1 |
| -Anti-CTLA-4 inhibitor -TLR agonist | Combo IO | S | 1.7 | 5 | Inactivating: PIK3R1 (c.1727_1729del), TP53 (c.581T > G), and PI3KR1 (c.1112del) Unknown functional significance: FBXW7 |
| -Anti-PD-1 inhibitor | Mono | E | 1.4 | 4 | Unknown functional significance: TP53 |
| -Anti-PD-1 inhibitor -Anti-PD-L1 inhibitor | Combo IO | E | 0.9 | 5 | Inactivating: PTEN (c.388C > G) and TP53 (c.659A > G) Unknown functional significance: PIK3R1, RNF43, and FBXW7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
How, J.A.; Jazaeri, A.A.; Fu, S.; Rodon Ahnert, J.; Gong, J.; Stephen, B.; Ferreira Dalla Pria, H.; Bhosale, P.; Johnson, A.; Yuan, Y.; et al. Clinical Outcomes of Patients with Recurrent Microsatellite-Stable Endometrial Cancer in Early-Phase Immunotherapy Clinical Trials. Cancers 2022, 14, 3695. https://doi.org/10.3390/cancers14153695
How JA, Jazaeri AA, Fu S, Rodon Ahnert J, Gong J, Stephen B, Ferreira Dalla Pria H, Bhosale P, Johnson A, Yuan Y, et al. Clinical Outcomes of Patients with Recurrent Microsatellite-Stable Endometrial Cancer in Early-Phase Immunotherapy Clinical Trials. Cancers. 2022; 14(15):3695. https://doi.org/10.3390/cancers14153695
Chicago/Turabian StyleHow, Jeffrey A., Amir A. Jazaeri, Siqing Fu, Jordi Rodon Ahnert, Jing Gong, Bettzy Stephen, Hanna Ferreira Dalla Pria, Priya Bhosale, Amber Johnson, Ying Yuan, and et al. 2022. "Clinical Outcomes of Patients with Recurrent Microsatellite-Stable Endometrial Cancer in Early-Phase Immunotherapy Clinical Trials" Cancers 14, no. 15: 3695. https://doi.org/10.3390/cancers14153695