
Lung Cancer in the Course of COPD-Emerging Problems Today



Abstract
Simple Summary
Abstract
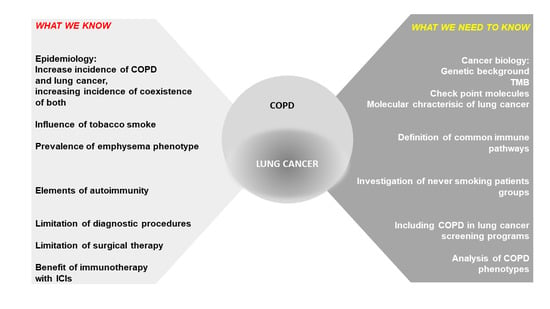
1. Introduction
Epidemiology
2. Pathogenesis and Common Pathways
2.1. Inflammation
2.2. Immunity
2.2.1. Lymphocytes
2.2.2. Autoimmunity
2.3. Genetic Candidates
2.4. Epigenetics
3. Going to the Clinic
3.1. Comorbidity
3.2. COPD Is Not a Homogenic Disease
3.3. Difficulties in Diagnosis
3.4. COPD Influence Treatment of Lung Cancer
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- West, R. Tobacco smoking: Health impact, prevalence, correlates and interventions. Psychol. Health 2017, 32, 1018–1036. [Google Scholar] [CrossRef]
- World Health Organization. WHO Global Report on Trends in Prevalence of Tobacco Use 2000–2025; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar]
- Mu, L.; Liu, L.; Niu, R.; Zhao, B.; Shi, J.; Li, Y.; Swanson, M.; Scheider, W.; Su, J.; Chang, S.-C.; et al. Indoor air pollution and risk of lung cancer among Chinese female non-smokers. Cancer Causes Control 2013, 24, 439–450. [Google Scholar] [CrossRef]
- Turner, M.C.; Andersen, Z.J.; Baccarelli, A.; Diver, W.R.; Gapstur, S.M.; Pope, C.A., 3rd; Prada, D.; Samet, J.; Thurston, G.; Cohen, A. Outdoor air pollution and cancer: An overview of the current evidence and public health recommendations. CA Cancer J. Clin. 2020, 70, 460–479. [Google Scholar] [CrossRef]
- Devesa, S.S.; Blot, W.J.; Fraumeni, J.F., Jr. Declining lung cancer rates among young men and women in the United States: A cohort analysis. J. Natl. Cancer Inst. 1989, 81, 1568–1571. [Google Scholar] [CrossRef]
- Lortet-Tieulent, J.; Renteria, E.; Sharp, L.; Weiderpass, E.; Comber, H.; Baas, P.; Bray, F.; Coebergh, J.W.; Soerjomataram, I. Convergence of decreasing male and increasing female incidence rates in major tobacco-related cancers in Europe in 1988–2010. Eur. J. Cancer 2015, 51, 1144–1163. [Google Scholar] [CrossRef]
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Niksic, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): Analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef]
- GBD 2017 DALYs; Hale Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 359 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1859–1922. [Google Scholar] [CrossRef]
- Adeloye, D.; Chua, S.; Lee, C.; Basquill, C.; Papana, A.; Theodoratou, E.; Nair, H.; Gasevic, D.; Sridhar, D.; Campbell, H.; et al. Global and regional estimates of COPD prevalence: Systematic review and meta-analysis. J. Glob. Health 2015, 5, 020415. [Google Scholar] [CrossRef]
- WHO. Global Health Estimates. Available online: https://www.who.int/news-room/fact-sheets/detail/chronic-obstructive-pulmonary-disease-(copd) (accessed on 20 May 2022).
- Durham, A.L.; Adcock, I.M. The relationship between COPD and lung cancer. Lung Cancer 2015, 90, 121–127. [Google Scholar] [CrossRef]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef]
- Terzikhan, N.; Verhamme, K.M.; Hofman, A.; Stricker, B.H.; Brusselle, G.G.; Lahousse, L. Prevalence and incidence of COPD in smokers and non-smokers: The Rotterdam Study. Eur. J. Epidemiol. 2016, 31, 785–792. [Google Scholar] [CrossRef]
- Young, R.P.; Hopkins, R.J.; Gamble, G.D.; Etzel, C.; El-Zein, R.; Crapo, J.D. Genetic evidence linking lung cancer and COPD: A new perspective. Appl. Clin. Genet. 2011, 4, 99–111. [Google Scholar] [CrossRef]
- Available online: https://goldcopd.org/ (accessed on 20 May 2022).
- Hodge, S.; Hodge, G.; Holmes, M.; Reynolds, P.N. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur. Respir. J. 2005, 25, 447–454. [Google Scholar] [CrossRef]
- Domagala-Kulawik, J.; Maskey-Warzechowska, M.; Kraszewska, I.; Chazan, R. The cellular composition and macrophage phenotype in induced sputum in smokers and ex-smokers with COPD. Chest 2003, 123, 1054–1059. [Google Scholar] [CrossRef]
- Smith, C.J.; Livingston, S.D.; Doolittle, D.J. An international literature survey of “IARC Group I carcinogens” reported in mainstream cigarette smoke. Food Chem. Toxicol. 1997, 35, 1107–1130. [Google Scholar] [CrossRef]
- Edwards, D. Immunological effects of tobacco smoking in “healthy” smokers. COPD 2009, 6, 48–58. [Google Scholar] [CrossRef]
- Pandey, K.C.; De, S.; Mishra, P.K. Role of Proteases in Chronic Obstructive Pulmonary Disease. Front. Pharmacol. 2017, 8, 512. [Google Scholar] [CrossRef]
- Linder, R.; Ronmark, E.; Pourazar, J.; Behndig, A.; Blomberg, A.; Lindberg, A. Serum metalloproteinase-9 is related to COPD severity and symptoms—Cross-sectional data from a population based cohort-study. Respir. Res. 2015, 16, 28. [Google Scholar] [CrossRef]
- Sorroche, P.B.; Fernandez Acquier, M.; Lopez Jove, O.; Giugno, E.; Pace, S.; Livellara, B.; Legal, S.; Oyhamburu, J.; Saez, M.S. Alpha-1 Antitrypsin Deficiency in COPD Patients: A Cross-Sectional Study. Arch. Bronconeumol. 2015, 51, 539–543. [Google Scholar] [CrossRef]
- Hecht, S.S. Lung carcinogenesis by tobacco smoke. Int. J. Cancer 2012, 131, 2724–2732. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.M.; Pikor, L.A.; Kung, S.H.; Hamilton, M.J.; Lam, S.; Lam, W.L.; Bennewith, K.L. Macrophages, Inflammation, and Lung Cancer. Am. J. Respir. Crit. Care Med. 2016, 193, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Aloe, C.; Wang, H.; Vlahos, R.; Irving, L.; Steinfort, D.; Bozinovski, S. Emerging and multifaceted role of neutrophils in lung cancer. Transl. Lung Cancer Res. 2021, 10, 2806–2818. [Google Scholar] [CrossRef] [PubMed]
- Church, D.F.; Pryor, W.A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 1985, 64, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Tergaonkar, V. NFkappaB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis 2009, 14, 348–363. [Google Scholar] [CrossRef]
- Domagala-Kulawik, J. New Frontiers for Molecular Pathology. Front. Med. 2019, 6, 284. [Google Scholar] [CrossRef]
- Kachuri, L.; Johansson, M.; Rashkin, S.R.; Graff, R.E.; Bosse, Y.; Manem, V.; Caporaso, N.E.; Landi, M.T.; Christiani, D.C.; Vineis, P.; et al. Immune-mediated genetic pathways resulting in pulmonary function impairment increase lung cancer susceptibility. Nat. Commun. 2020, 11, 27. [Google Scholar] [CrossRef]
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033. [Google Scholar] [CrossRef]
- Domagala-Kulawik, J. Effects of cigarette smoke on the lung and systemic immunity. J. Physiol. Pharmacol. 2008, 59 (Suppl. 6), 19–34. [Google Scholar]
- Collier, J.L.; Weiss, S.A.; Pauken, K.E.; Sen, D.R.; Sharpe, A.H. Not-so-opposite ends of the spectrum: CD8(+) T cell dysfunction across chronic infection, cancer and autoimmunity. Nat. Immunol. 2021, 22, 809–819. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.M. Transforming growth factor beta: The good, the bad, and the ugly. J. Exp. Med. 1994, 180, 1587–1590. [Google Scholar] [CrossRef]
- Hodge, S.J.; Hodge, G.L.; Reynolds, P.N.; Scicchitano, R.; Holmes, M. Increased production of TGF-beta and apoptosis of T lymphocytes isolated from peripheral blood in COPD. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L492–L499. [Google Scholar] [CrossRef]
- Curtis, J.L.; Freeman, C.M.; Hogg, J.C. The immunopathogenesis of chronic obstructive pulmonary disease: Insights from recent research. Proc. Am. Thorac. Soc. 2007, 4, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Biton, J.; Ouakrim, H.; Dechartres, A.; Alifano, M.; Mansuet-Lupo, A.; Si, H.; Halpin, R.; Creasy, T.; Bantsimba-Malanda, C.; Arrondeau, J.; et al. Impaired Tumor-Infiltrating T Cells in Patients with Chronic Obstructive Pulmonary Disease Impact Lung Cancer Response to PD-1 Blockade. Am. J. Respir. Crit. Care Med. 2018, 198, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Anderson, G.P.; Fageras, M.; Belvisi, M.G. Chronic lung diseases: Prospects for regeneration and repair. Eur. Respir. Rev. 2021, 30, 200213. [Google Scholar] [CrossRef]
- Uller, L.; Persson, C.G.; Erjefalt, J.S. Resolution of airway disease: Removal of inflammatory cells through apoptosis, egression or both? Trends Pharm. Sci. 2006, 27, 461–466. [Google Scholar] [CrossRef]
- Domagala-Kulawik, J.; Hoser, G.; Dabrowska, M.; Chazan, R. Increased proportion of Fas positive CD8+ cells in peripheral blood of patients with COPD. Respir. Med. 2007, 101, 1338–1343. [Google Scholar] [CrossRef][Green Version]
- Hoser, G.; Wasilewska, D.; Domagala-Kulawik, J. Expression of Fas receptor on peripheral blood lymphocytes from patients with non-small cell lung cancer. Folia Histochem. Cytobiol. 2004, 42, 249–252. [Google Scholar]
- Majo, J.; Ghezzo, H.; Cosio, M.G. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir. J. 2001, 17, 946–953. [Google Scholar] [CrossRef]
- Kerr, K.M. Pulmonary preinvasive neoplasia. J. Clin. Pathol. 2001, 54, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Casoni, G.; Caramori, G.; Guzzinati, I.; Boschetto, P.; Ravenna, F.; Calia, N.; Petruzzelli, S.; Corbetta, L.; Cavallesco, G.; et al. COPD increases the risk of squamous histological subtype in smokers who develop non-small cell lung carcinoma. Thorax 2004, 59, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Dunmore, R.; Camelo, A.; Da Silva, C.A.; Gustavsson, M.J.; Habiel, D.M.; Hackett, T.L.; Hogaboam, C.M.; Sleeman, M.A.; Knight, D.A. Acute cigarette smoke exposure activates apoptotic and inflammatory programs but a second stimulus is required to induce epithelial to mesenchymal transition in COPD epithelium. Respir. Res. 2017, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Caramori, G.; Capelli, A.; Gnemmi, I.; Ricciardolo, F.L.; Oates, T.; Donner, C.; Chung, K.F.; Barnes, P.; Adcock, I. STAT4 activation in smokers and patients with chronic obstructive pulmonary disease. Eur. Respir. J. 2004, 24, 78–85. [Google Scholar] [CrossRef]
- Sullivan, A.K.; Simonian, P.L.; Falta, M.T.; Mitchell, J.D.; Cosgrove, G.P.; Brown, K.K.; Kotzin, B.L.; Voelkel, N.F.; Fontenot, A.P. Oligoclonal CD4+ T cells in the lungs of patients with severe emphysema. Am. J. Respir. Crit. Care Med. 2005, 172, 590–596. [Google Scholar] [CrossRef]
- Taraseviciene-Stewart, L.; Douglas, I.S.; Nana-Sinkam, P.S.; Lee, J.D.; Tuder, R.M.; Nicolls, M.R.; Voelkel, N.F. Is alveolar destruction and emphysema in chronic obstructive pulmonary disease an immune disease? Proc. Am. Thorac. Soc. 2006, 3, 687–690. [Google Scholar] [CrossRef]
- Saetta, M. Airway inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999, 160, S17–S20. [Google Scholar] [CrossRef]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef]
- Wilkinson, T.M.A. Immune checkpoints in chronic obstructive pulmonary disease. Eur. Respir. Rev. 2017, 26, 170045. [Google Scholar] [CrossRef]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef]
- Mark, N.M.; Kargl, J.; Busch, S.E.; Yang, G.H.Y.; Metz, H.E.; Zhang, H.; Hubbard, J.J.; Pipavath, S.N.J.; Madtes, D.K.; Houghton, A.M. Chronic Obstructive Pulmonary Disease Alters Immune Cell Composition and Immune Checkpoint Inhibitor Efficacy in Non-Small Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 2018, 197, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Byrne, R.; Todd, I.; Tighe, P.J.; Fairclough, L.C. Autoantibodies in chronic obstructive pulmonary disease: A systematic review. Immunol. Lett. 2019, 214, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Taraseviciene-Stewart, L.; Voelkel, N.F. Molecular pathogenesis of emphysema. J. Clin. Investig. 2008, 118, 394–402. [Google Scholar] [CrossRef]
- Stefanska, A.M.; Walsh, P.T. Chronic obstructive pulmonary disease: Evidence for an autoimmune component. Cell Mol. Immunol. 2009, 6, 81–86. [Google Scholar] [CrossRef]
- Zeneyedpour, L.; Dekker, L.J.M.; van Sten-van, T.H.J.J.M.; Burgers, P.C.; Ten Hacken, N.H.T.; Luider, T.M. Neoantigens in Chronic Obstructive Pulmonary Disease and Lung Cancer: A Point of View. Proteom. Clin. Appl. 2019, 13, e1800093. [Google Scholar] [CrossRef] [PubMed]
- Christenson, S.A.; Smith, B.M.; Bafadhel, M.; Putcha, N. Chronic obstructive pulmonary disease. Lancet 2022, 399, 2227–2242. [Google Scholar] [CrossRef]
- Caramori, G.; Ruggeri, P.; Di Stefano, A.; Mumby, S.; Girbino, G.; Adcock, I.M.; Kirkham, P. Autoimmunity and COPD: Clinical Implications. Chest 2018, 153, 1424–1431. [Google Scholar] [CrossRef]
- Bocian, K.; Kiernozek, E.; Domagala-Kulawik, J.; Korczak-Kowalska, G.; Stelmaszczyk-Emmel, A.; Drela, N. Expanding Diversity and Common Goal of Regulatory T and B Cells. I: Origin, Phenotype, Mechanisms. Arch. Immunol. Ther. Exp. 2017, 65, 501–520. [Google Scholar] [CrossRef]
- Booth, N.J.; McQuaid, A.J.; Sobande, T.; Kissane, S.; Agius, E.; Jackson, S.E.; Salmon, M.; Falciani, F.; Yong, K.; Rustin, M.H.; et al. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J. Immunol. 2010, 184, 4317–4326. [Google Scholar] [CrossRef]
- Singh, R.; Alape, D.; de Lima, A.; Ascanio, J.; Majid, A.; Gangadharan, S.P. Regulatory T Cells in Respiratory Health and Diseases. Pulm. Med. 2019, 2019, 1907807. [Google Scholar] [CrossRef]
- Domagala-Kulawik, J.; Hoser, G.; Dabrowska, M.; Safianowska, A.; Chazan, R. CD4+/CD25+ cells in systemic inflammation in COPD. Scand. J. Immunol. 2011, 73, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Sun, Y. Role of Regulatory T Cells in Disturbed Immune Homeostasis in Patients with Chronic Obstructive Pulmonary Disease. Front. Immunol. 2020, 11, 723. [Google Scholar] [CrossRef] [PubMed]
- Shim, C.H.; Cho, S.; Shin, Y.M.; Choi, J.M. Emerging role of bystander T cell activation in autoimmune diseases. BMB Rep. 2022, 55, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Caramori, G.; Gnemmi, I.; Contoli, M.; Bristot, L.; Capelli, A.; Ricciardolo, F.L.M.; Magno, F.; D’Anna, S.E.; Zanini, A.; et al. Association of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPD. Thorax 2009, 64, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Mirabolfathinejad, S.G.; Katta, H.; Cumpian, A.M.; Gong, L.; Caetano, M.S.; Moghaddam, S.J.; Dong, C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 5664–5669. [Google Scholar] [CrossRef] [PubMed]
- Sakowska, J.; Arcimowicz, L.; Jankowiak, M.; Papak, I.; Markiewicz, A.; Dziubek, K.; Kurkowiak, M.; Kote, S.; Kaźmierczak-Siedlecka, K.; Połom, K.; et al. Autoimmunity and Cancer-Two Sides of the Same Coin. Front. Immunol. 2022, 13, 793234. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Y.; Han, J.; Yang, M.; Zhu, J.; Jin, T. Transitional B cells involved in autoimmunity and their impact on neuroimmunological diseases. J. Transl. Med. 2020, 18, 131. [Google Scholar] [CrossRef]
- Benvenuto, M.; Mattera, R.; Masuelli, L.; Tresoldi, I.; Giganti, M.G.; Frajese, G.V.; Manzari, V.; Modesti, A.; Bei, R. The crossroads between cancer immunity and autoimmunity: Antibodies to self antigens. Front. Biosci. 2017, 22, 1289–1329. [Google Scholar]
- Li, C.M.; Chen, Z. Autoimmunity as an Etiological Factor of Cancer: The Transformative Potential of Chronic Type 2 Inflammation. Front. Cell Dev. Biol. 2021, 9, 664305. [Google Scholar] [CrossRef]
- Hung, R.J.; McKay, J.D.; Gaborieau, V.; Boffetta, P.; Hashibe, M.; Zaridze, D.; Mukeria, A.; Szeszenia-Dabrowska, N.; Lissowska, J.; Rudnai, P.; et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 2008, 452, 633–637. [Google Scholar] [CrossRef]
- Saccone, N.L.; Wang, J.C.; Breslau, N.; Johnson, E.O.; Hatsukami, D.; Saccone, S.F.; Grucza, R.A.; Sun, L.; Duan, W.; Budde, J.; et al. The CHRNA5-CHRNA3-CHRNB4 nicotinic receptor subunit gene cluster affects risk for nicotine dependence in African-Americans and in European-Americans. Cancer Res. 2009, 69, 6848–6856. [Google Scholar] [CrossRef] [PubMed]
- Le Marchand, L.; Derby, K.S.; Murphy, S.E.; Hecht, S.S.; Hatsukami, D.; Carmella, S.G.; Tiirikainen, M.; Wang, H. Smokers with the CHRNA lung cancer-associated variants are exposed to higher levels of nicotine equivalents and a carcinogenic tobacco-specific nitrosamine. Cancer Res. 2008, 68, 9137–9140. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Tyndale, R.F.; Lerman, C. Nicotine dependence pharmacogenetics: Role of genetic variation in nicotine-metabolizing enzymes. J. Neurogenet. 2009, 23, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, C.A.; Dong, Q.; Wei, Q.; Amos, C.I.; Spitz, M.R.; Tyndale, R.F. Relationship between CYP2A6 and CHRNA5-CHRNA3-CHRNB4 variation and smoking behaviors and lung cancer risk. J. Natl. Cancer Inst. 2011, 103, 1342–1346. [Google Scholar] [CrossRef]
- Young, R.P.; Hopkins, R.J.; Hay, B.A.; Whittington, C.F.; Epton, M.J.; Gamble, G.D. FAM13A locus in COPD is independently associated with lung cancer—Evidence of a molecular genetic link between COPD and lung cancer. Appl. Clin. Genet. 2011, 4, 1–10. [Google Scholar] [CrossRef]
- Corvol, H.; Hodges, C.A.; Drumm, M.L.; Guillot, L. Moving beyond genetics: Is FAM13A a major biological contributor in lung physiology and chronic lung diseases? J. Med. Genet. 2014, 51, 646–649. [Google Scholar] [CrossRef]
- Young, R.P.; Whittington, C.F.; Hopkins, R.J.; Hay, B.A.; Epton, M.J.; Black, P.N.; Gamble, G.D. Chromosome 4q31 locus in COPD is also associated with lung cancer. Eur. Respir. J. 2010, 36, 1375–1382. [Google Scholar] [CrossRef]
- Shi, W.; Chen, F.; Cardoso, W.V. Mechanisms of lung development: Contribution to adult lung disease and relevance to chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2009, 6, 558–563. [Google Scholar] [CrossRef]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef]
- Lemjabbar-Alaoui, H.; Dasari, V.; Sidhu, S.S.; Mengistab, A.; Finkbeiner, W.; Gallup, M.; Basbaum, C. Wnt and Hedgehog are critical mediators of cigarette smoke-induced lung cancer. PLoS ONE 2006, 1, e93. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Launay, J.M.; Del Pino, M.; Chironi, G.; Callebert, J.; Peoc’h, K.; Megnien, J.L.; Mallet, J.; Simon, A.; Rendu, F. Smoking induces long-lasting effects through a monoamine-oxidase epigenetic regulation. PLoS ONE 2009, 4, e7959. [Google Scholar] [CrossRef] [PubMed]
- Tessema, M.; Yingling, C.M.; Picchi, M.A.; Wu, G.; Liu, Y.; Weissfeld, J.L.; Siegfried, J.M.; Tesfaigzi, Y.; Belinsky, S.A. Epigenetic Repression of CCDC37 and MAP1B Links Chronic Obstructive Pulmonary Disease to Lung Cancer. J. Thorac. Oncol. 2015, 10, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Baccarelli, A.; Carey, V.J.; Boutaoui, N.; Bacherman, H.; Klanderman, B.; Rennard, S.; Agusti, A.; Anderson, W.; Lomas, D.A.; et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am. J. Respir. Crit. Care Med. 2012, 185, 373–381. [Google Scholar] [CrossRef]
- Toden, S.; Zumwalt, T.J.; Goel, A. Non-coding RNAs and potential therapeutic targeting in cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188491. [Google Scholar] [CrossRef] [PubMed]
- Schetter, A.J.; Heegaard, N.H.; Harris, C.C. Inflammation and cancer: Interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 2010, 31, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.E.; Perry, M.M.; Moschos, S.A.; Larner-Svensson, H.M.; Lindsay, M.A. Role of miRNA-146a in the regulation of the innate immune response and cancer. Biochem. Soc. Trans. 2008, 36 Pt 6, 1211–1215. [Google Scholar] [CrossRef]
- Fathinavid, A.; Ghobadi, M.Z.; Najafi, A.; Masoudi-Nejad, A. Identification of common microRNA between COPD and non-small cell lung cancer through pathway enrichment analysis. BMC Genom. Data 2021, 22, 41. [Google Scholar] [CrossRef]
- Grose, D.; Morrison, D.S.; Devereux, G.; Jones, R.; Sharma, D.; Selby, C.; Docherty, K.; McIntosh, D.; Louden, G.; Nicolson, M.; et al. Comorbidities in lung cancer: Prevalence, severity and links with socioeconomic status and treatment. Postgrad. Med. J. 2014, 90, 305–310. [Google Scholar] [CrossRef]
- Negewo, N.A.; McDonald, V.M.; Gibson, P.G. Comorbidity in chronic obstructive pulmonary disease. Respir. Investig. 2015, 53, 249–258. [Google Scholar] [CrossRef]
- Sekine, Y.; Katsura, H.; Koh, E.; Hiroshima, K.; Fujisawa, T. Early detection of COPD is important for lung cancer surveillance. Eur. Respir. J. 2012, 39, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Dutkowska, A.E.; Antczak, A. Comorbidities in lung cancer. Pneumonol. Alergol. Pol. 2016, 84, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Van Eeden, S.; Leipsic, J.; Paul Man, S.F.; Sin, D.D. The relationship between lung inflammation and cardiovascular disease. Am. J. Respir. Crit. Care Med. 2012, 186, 11–16. [Google Scholar] [CrossRef] [PubMed]
- King, P.T. Inflammation in chronic obstructive pulmonary disease and its role in cardiovascular disease and lung cancer. Clin. Transl. Med. 2015, 4, 68. [Google Scholar] [CrossRef]
- Finkelstein, J.; Cha, E.; Scharf, S.M. Chronic obstructive pulmonary disease as an independent risk factor for cardiovascular morbidity. Int. J. Chron. Obs. Pulmon. Dis. 2009, 4, 337–349. [Google Scholar] [CrossRef]
- Peinado, V.I.; Barbera, J.A.; Ramirez, J.; Gomez, F.P.; Roca, J.; Jover, L.; Gimferrer, J.M.; Rodriguez-Roisin, R. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am. J. Physiol. 1998, 274, L908–L913. [Google Scholar] [CrossRef]
- Tillie-Leblond, I.; Marquette, C.H.; Perez, T.; Scherpereel, A.; Zanetti, C.; Tonnel, A.B.; Remy-Jardin, M. Pulmonary embolism in patients with unexplained exacerbation of chronic obstructive pulmonary disease: Prevalence and risk factors. Ann. Intern. Med. 2006, 144, 390–396. [Google Scholar] [CrossRef]
- Tamagawa, E.; van Eeden, S.F. Impaired lung function and risk for stroke: Role of the systemic inflammation response? Chest 2006, 130, 1631–1633. [Google Scholar] [CrossRef]
- Vaidyula, V.R.; Criner, G.J.; Grabianowski, C.; Rao, A.K. Circulating tissue factor procoagulant activity is elevated in stable moderate to severe chronic obstructive pulmonary disease. Thromb. Res. 2009, 124, 259–261. [Google Scholar] [CrossRef]
- Undas, A.; Kaczmarek, P.; Sladek, K.; Stepien, E.; Skucha, W.; Rzeszutko, M.; Gorkiewicz-Kot, I.; Tracz, W. Fibrin clot properties are altered in patients with chronic obstructive pulmonary disease. Beneficial effects of simvastatin treatment. Thromb. Haemost. 2009, 102, 1176–1182. [Google Scholar]
- Au, D.H.; Udris, E.M.; Fihn, S.D.; McDonell, M.B.; Curtis, J.R. Differences in health care utilization at the end of life among patients with chronic obstructive pulmonary disease and patients with lung cancer. Arch. Intern. Med. 2006, 166, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Celli, B.R.; Agusti, A. COPD: Time to improve its taxonomy? ERJ Open Res. 2018, 4, 00132-2017. [Google Scholar] [CrossRef] [PubMed]
- Corlateanu, A.; Mendez, Y.; Wang, Y.; Garnica, R.J.A.; Botnaru, V.; Siafakas, N. Chronic obstructive pulmonary disease and phenotypes: A state-of-the-art. Pulmonology 2020, 26, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Vestbo, J. COPD: Definition and phenotypes. Clin. Chest Med. 2014, 35, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.; Han, M.K.; Vance, G.B.; Make, B.J.; Newell, J.D.; Hokanson, J.E.; Hersh, C.P.; Stinson, D.; Silverman, E.K.; Criner, G.J. The chronic bronchitic phenotype of COPD: An analysis of the COPDGene Study. Chest 2011, 140, 626–633. [Google Scholar] [CrossRef]
- Seemungal, T.A.; Wilkinson, T.M.; Hurst, J.R.; Perera, W.R.; Sapsford, R.J.; Wedzicha, J.A. Long-term erythromycin therapy is associated with decreased chronic obstructive pulmonary disease exacerbations. Am. J. Respir. Crit. Care Med. 2008, 178, 1139–1147. [Google Scholar] [CrossRef]
- Kim, V.; Criner, G.J. The chronic bronchitis phenotype in chronic obstructive pulmonary disease: Features and implications. Curr. Opin. Pulm. Med. 2015, 21, 133–141. [Google Scholar] [CrossRef]
- Gonzalez, J.; Henschke, C.I.; Yankelevitz, D.F.; Seijo, L.M.; Reeves, A.P.; Yip, R.; Xie, Y.; Chung, M.; Sánchez-Salcedo, P.; Alcaide, A.B.; et al. Emphysema phenotypes and lung cancer risk. PLoS ONE 2019, 14, e0219187. [Google Scholar] [CrossRef]
- Wang, W.; Xie, M.; Dou, S.; Cui, L.; Zheng, C.; Xiao, W. The link between chronic obstructive pulmonary disease phenotypes and histological subtypes of lung cancer: A case-control study. Int. J. Chron. Obs. Pulm. Dis. 2018, 13, 1167–1175. [Google Scholar] [CrossRef]
- Mirza, S.; Benzo, R. Chronic Obstructive Pulmonary Disease Phenotypes: Implications for Care. Mayo Clin. Proc. 2017, 92, 1104–1112. [Google Scholar] [CrossRef]
- Soler-Cataluna, J.J.; Cosio, B.; Izquierdo, J.L.; Lopez-Campos, J.L.; Marin, J.M.; Aguero, R.; Baloira, A.; Carrizo, S.; Esteban, C.; Galdiz, J.B.; et al. Consensus document on the overlap phenotype COPD-asthma in COPD. Arch. Bronconeumol. 2012, 48, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Halpin, D.M.; Decramer, M.; Celli, B.; Kesten, S.; Liu, D.; Tashkin, D.P. Exacerbation frequency and course of COPD. Int. J. Chron. Obs. Pulm. Dis. 2012, 7, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.J.; Kneppers, A.E.; van de Bool, C.; Langen, R.C.; Schols, A.M. Cachexia in chronic obstructive pulmonary disease: New insights and therapeutic perspective. J. Cachexia Sarcopenia Muscle 2016, 7, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Vestbo, J.; Prescott, E.; Almdal, T.; Dahl, M.; Nordestgaard, B.G.; Andersen, T.; Sørensen, T.I.A.; Lange, P. Body mass, fat-free body mass, and prognosis in patients with chronic obstructive pulmonary disease from a random population sample: Findings from the Copenhagen City Heart Study. Am. J. Respir. Crit. Care Med. 2006, 173, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Romme, E.A.; Murchison, J.T.; Phang, K.F.; Jansen, F.H.; Rutten, E.P.; Wouters, E.F.; Smeenk, F.W.; Van Beek, E.J.; MacNee, W. Bone attenuation on routine chest CT correlates with bone mineral density on DXA in patients with COPD. J. Bone Miner. Res. 2012, 27, 2338–2343. [Google Scholar] [CrossRef]
- Mittal, N.; Raj, R.; Islam, E.A.; Nugent, K. The Frequency of Frailty in Ambulatory Patients with Chronic Lung Diseases. J. Prim. Care Community Health 2016, 7, 10–15. [Google Scholar] [CrossRef]
- Pooler, A.; Beech, R. Examining the relationship between anxiety and depression and exacerbations of COPD which result in hospital admission: A systematic review. Int. J. Chron. Obs. Pulm. Dis. 2014, 9, 315–330. [Google Scholar] [CrossRef]
- Hurst, J.R.; Elborn, J.S.; De Soyza, A.; BRONCH-UK Consortium. COPD-bronchiectasis overlap syndrome. Eur. Respir. J. 2015, 45, 310–313. [Google Scholar] [CrossRef]
- Fishman, A.; Martinez, F.; Naunheim, K.; Piantadosi, S.; Wise, R.; Ries, A.; Weinmann, G.; Wood, D.E.; National Emphysema Treatment Trial Research Group. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N. Engl. J. Med. 2003, 348, 2059–2073. [Google Scholar]
- Pinto, L.M.; Alghamdi, M.; Benedetti, A.; Zaihra, T.; Landry, T.; Bourbeau, J. Derivation and validation of clinical phenotypes for COPD: A systematic review. Respir. Res. 2015, 16, 50. [Google Scholar] [CrossRef]
- Mouronte-Roibas, C.; Leiro-Fernandez, V.; Fernandez-Villar, A.; Botana-Rial, M.; Ramos-Hernandez, C.; Ruano-Ravina, A. COPD, emphysema and the onset of lung cancer. A systematic review. Cancer Lett. 2016, 382, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Tubio-Perez, R.A.; Torres-Duran, M.; Perez-Rios, M.; Fernandez-Villar, A.; Ruano-Ravina, A. Lung emphysema and lung cancer: What do we know about it? Ann. Transl. Med. 2020, 8, 1471. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.R.; Caldeira, J.N.; Rodrigues, C.; Figueiredo, A.; Barata, F. Lung cancer screening in clinical practice: Identification of high-risk chronic obstructive pulmonary disease patients. Rev. Assoc. Med. Bras. 2022, 68, 502–506. [Google Scholar] [CrossRef] [PubMed]
- de-Torres, J.P.; Wilson, D.O.; Sanchez-Salcedo, P.; Weissfeld, J.L.; Berto, J.; Campo, A.; Alcaide, A.B.; García-Granero, M.; Celli, B.R.; Zulueta, J.J. Lung cancer in patients with chronic obstructive pulmonary disease. Development and validation of the COPD Lung Cancer Screening Score. Am. J. Respir. Crit. Care Med. 2015, 191, 285–291. [Google Scholar] [CrossRef]
- Postmus, P.E.; Kerr, K.M.; Oudkerk, M.; Senan, S.; Waller, D.A.; Vansteenkiste, J.; Escriu, C.; Peters, S.; ESMO Guidelines Committee. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28 (Suppl. 4), iv1–iv21. [Google Scholar] [CrossRef]
- Torchio, R.; Guglielmo, M.; Giardino, R.; Ardissone, F.; Ciacco, C.; Gulotta, C.; Veljkovic, A.; Bugiani, M. Exercise ventilatory inefficiency and mortality in patients with chronic obstructive pulmonary disease undergoing surgery for non-small-cell lung cancer. Eur. J. Cardiothorac. Surg. 2010, 38, 14–19. [Google Scholar] [CrossRef]
- Hashimoto, N.; Matsuzaki, A.; Okada, Y.; Imai, N.; Iwano, S.; Wakai, K.; Imaizumi, K.; Yokoi, K.; Hasegawa, Y. Clinical impact of prevalence and severity of COPD on the decision-making process for therapeutic management of lung cancer patients. BMC Pulm. Med. 2014, 14, 14. [Google Scholar] [CrossRef][Green Version]
- Brunelli, A.; Charloux, A.; Bolliger, C.T.; Rocco, G.; Sculier, J.P.; Varela, G.; Licker, M.; Ferguson, M.K.; Faivre-Finn, C.; Huber, R.M.; et al. The European Respiratory Society and European Society of Thoracic Surgeons clinical guidelines for evaluating fitness for radical treatment (surgery and chemoradiotherapy) in patients with lung cancer. Eur. J. Cardiothorac. Surg. 2009, 36, 181–184. [Google Scholar] [CrossRef]
- Bolliger, C.T.; Jordan, P.; Soler, M.; Stulz, P.; Gradel, E.; Skarvan, K.; Elsasser, S.; Gonon, M.; Wyser, C.; Tamm, M. Exercise capacity as a predictor of postoperative complications in lung resection candidates. Am. J. Respir. Crit. Care Med. 1995, 151, 1472–1480. [Google Scholar] [CrossRef]
- Pierce, R.J.; Copland, J.M.; Sharpe, K.; Barter, C.E. Preoperative risk evaluation for lung cancer resection: Predicted postoperative product as a predictor of surgical mortality. Am. J. Respir. Crit. Care Med. 1994, 150, 947–955. [Google Scholar] [CrossRef]
- Brunelli, A.; Al Refai, M.; Monteverde, M.; Sabbatini, A.; Xiume, F.; Fianchini, A. Predictors of early morbidity after major lung resection in patients with and without airflow limitation. Ann. Thorac. Surg. 2002, 74, 999–1003. [Google Scholar] [CrossRef]
- Edwards, J.G.; Duthie, D.J.; Waller, D.A. Lobar volume reduction surgery: A method of increasing the lung cancer resection rate in patients with emphysema. Thorax 2001, 56, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Brunelli, A.; Refai, M.; Salati, M.; Xiume, F.; Sabbatini, A. Predicted versus observed FEV1 and DLCO after major lung resection: A prospective evaluation at different postoperative periods. Ann. Thorac. Surg. 2007, 83, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Brunelli, A.; Xiume, F.; Refai, M.; Salati, M.; Marasco, R.; Sciarra, V.; Sabbatini, A. Evaluation of expiratory volume, diffusion capacity, and exercise tolerance following major lung resection: A prospective follow-up analysis. Chest 2007, 131, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. 4), iv192–iv237. [Google Scholar] [CrossRef]
- Chennamadhavuni, A.; Abushahin, L.; Jin, N.; Presley, C.J.; Manne, A. Risk Factors and Biomarkers for Immune-Related Adverse Events: A Practical Guide to Identifying High-Risk Patients and Rechallenging Immune Checkpoint Inhibitors. Front. Immunol. 2022, 13, 779691. [Google Scholar]
- Domagala-Kulawik, J.; Leszek, P.; Owczarek, W.; Rawa, T.; Stelmachowska-Banas, M.; Rutkowski, P. Immunotherapy of solid tumors: Safety of treatment. Pol. Arch. Intern. Med. 2020, 130, 766–778. [Google Scholar]


{kind=link}
{kind=link}
| Phenotype | Characteristic | Reference |
|---|---|---|
| Chronic bronchitis | Associated with an accelerated lung function decline and an increased risk of respiratory infections, macrolide antibiotics with anti-inflammatory properties and phosphodiesterase- 4 inhibitors have been used to decrease COPD exacerbations and may be beneficial in the treatment. | [108,109,110] |
| Emphysematous | Presence of emphysema confirmed on chest imaging, relative to non-emphysema-predominant phenotype of COPD, emphysema-predominant phenotype has a higher risk of squamous-cell carcinoma and small-cell lung cancer, especially the centrilobular phenotype of emphysema increase lung cancer risk. | [111,112] |
| Asthma-COPD-Overlap (ACO) | Persistent airflow limitation usually with increased variability in airflow, airway obstruction is usually incompletely reversible and patients have several features associated with asthma as eosinophilia in sputum, personal history of asthma, high total IgE, personal history of atopy, and inhaled corticosteroids is an important treatment. | [113,114] |
| Frequent exacerbator | Presence of frequent exacerbations of two or more per year, there is three-fold increase of mortality in this phenotype. | [115] |
| Emerging COPD phenotypes | ||
| Pulmonary cachexia phenotype | With BMI lower than 21 kg/m2, extra-pulmonary degenerative manifestations include osteoporosis and muscle wasting (relatively high in COPD: 15–40% depending on definition and disease stage), muscle wasting not only contributes to diminished skeletal muscle function, reduced exercise capacity, but is also a determinant of mortality in COPD, independent of airflow obstruction. | [116,117,118] |
| Physical frailty phonotype | With weakness, slowness, low-level of physical activity, self-reported exhaustion and unintentional loss of weight, worse airflow limitation and symptoms, pulmonary rehabilitation can be effective. | [119] |
| Emotional frailty phenotype | Characterise by anxiety, depression, fear of breathlessness and is associated with increased morbidity, mortality and hospitalisation, patients should be supported by cognitive behavioural therapy. | [120] |
| Overlap COPD and bronchiectases | Confirmation of bronchiectases in HRCT and definite COPD diagnosis, both diseases have similar symptoms, because of that proper diagnosis and differentiation is a challenge, treatment useful in COPD may not be widely effective in bronchiectasis and vice versa (like ICS). | [121] |
| Upper lobe-predominant emphysema | Diagnosed by CT findings consistent of predominant upper lobe emphysema, significant improvement after LVRS. | [122] |
| Fast decliner phenotype | With rapid decline of lung function and high mortality, these patients should be early referred to specialised centres for aggressive disease management, lung transplantation should be considered early. | [123] |
| The comorbidities or systemic phenotype | High comorbidities burden, predominantly cardiovascular and metabolic, patients have increased mortality risk and should be managed by multispecialist teams. | [123] |
| α1-antitrypsin deficiency (AATD) | Genetic disorder associated with early onset COPD, highly under-diagnosed condition, early diagnosis could prompt specific interventions such as smoking cessation, testing of family members, genetic counselling and use of replacement therapy. | [23] |
| No smoking COPD | Induced by biomass exposure, especially among women in East Asia, which is caused by high outdoor ambient air pollution and exposure to household burning of solid fuels for heating and cooking. | [4,5] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uliński, R.; Kwiecień, I.; Domagała-Kulawik, J. Lung Cancer in the Course of COPD-Emerging Problems Today. Cancers 2022, 14, 3819. https://doi.org/10.3390/cancers14153819
Uliński R, Kwiecień I, Domagała-Kulawik J. Lung Cancer in the Course of COPD-Emerging Problems Today. Cancers. 2022; 14(15):3819. https://doi.org/10.3390/cancers14153819
Chicago/Turabian StyleUliński, Robert, Iwona Kwiecień, and Joanna Domagała-Kulawik. 2022. "Lung Cancer in the Course of COPD-Emerging Problems Today" Cancers 14, no. 15: 3819. https://doi.org/10.3390/cancers14153819
APA StyleUliński, R., Kwiecień, I., & Domagała-Kulawik, J. (2022). Lung Cancer in the Course of COPD-Emerging Problems Today. Cancers, 14(15), 3819. https://doi.org/10.3390/cancers14153819