
Detection of Microsatellite Instability in Colonoscopic Biopsies and Postal Urine Samples from Lynch Syndrome Cancer Patients Using a Multiplex PCR Assay

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. DNA Isolation
2.3. Two-Step Multiplex PCR and MIP Amplification
2.4. Sequencing and MSI Analysis
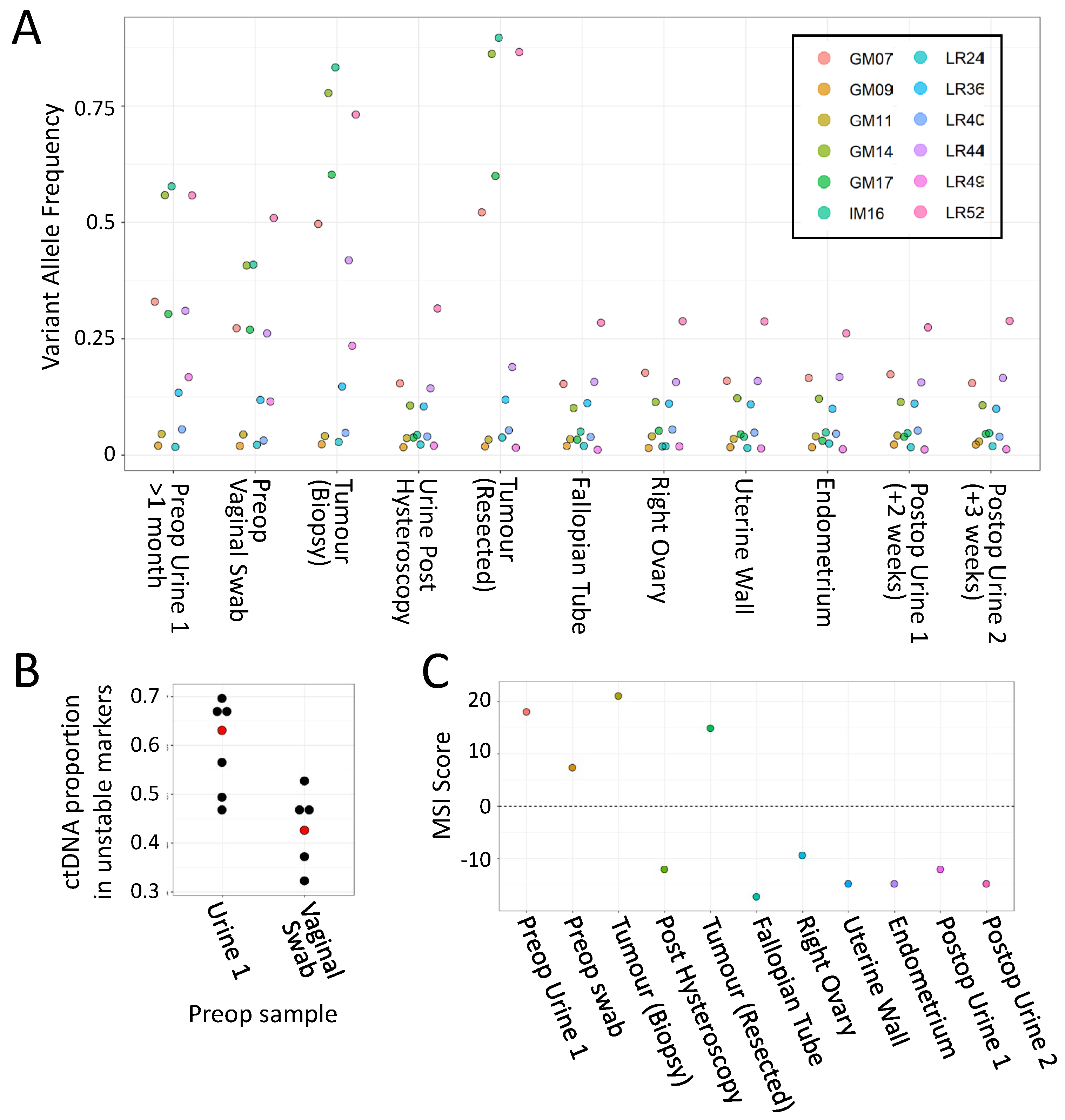
3. Results
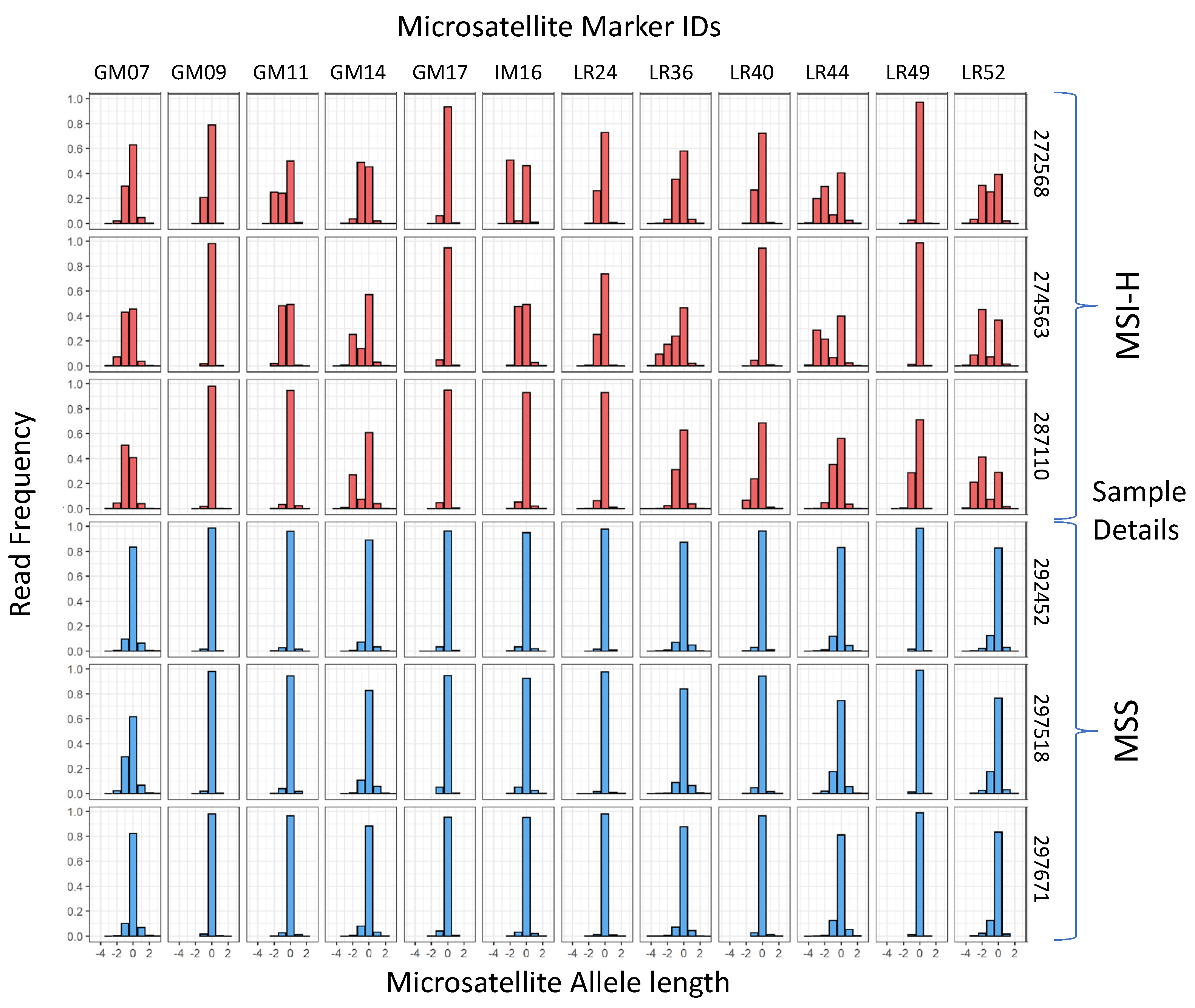
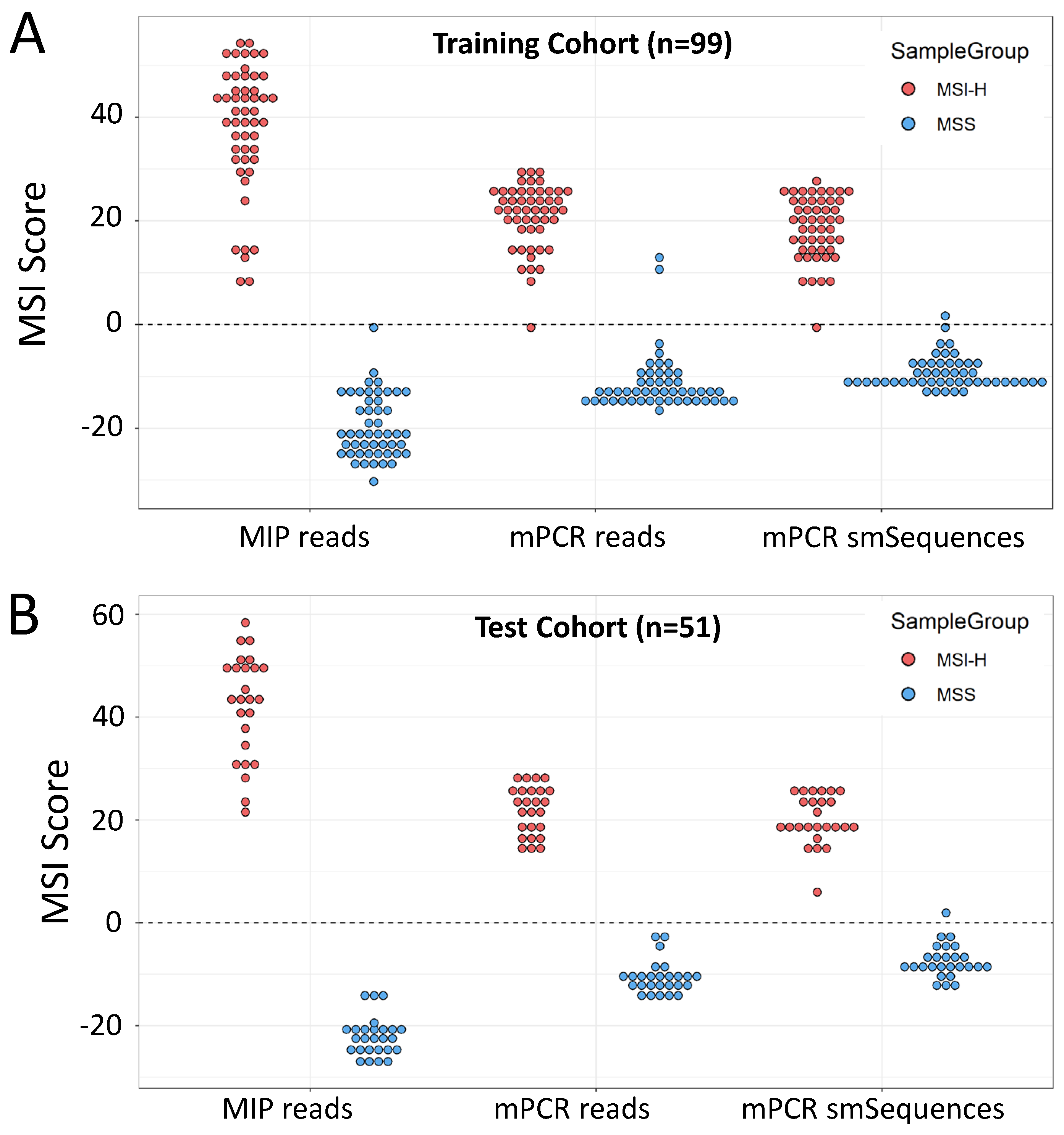
3.1. mPCR Classifies CRCs with 96–100% Sensitivity and Specificity
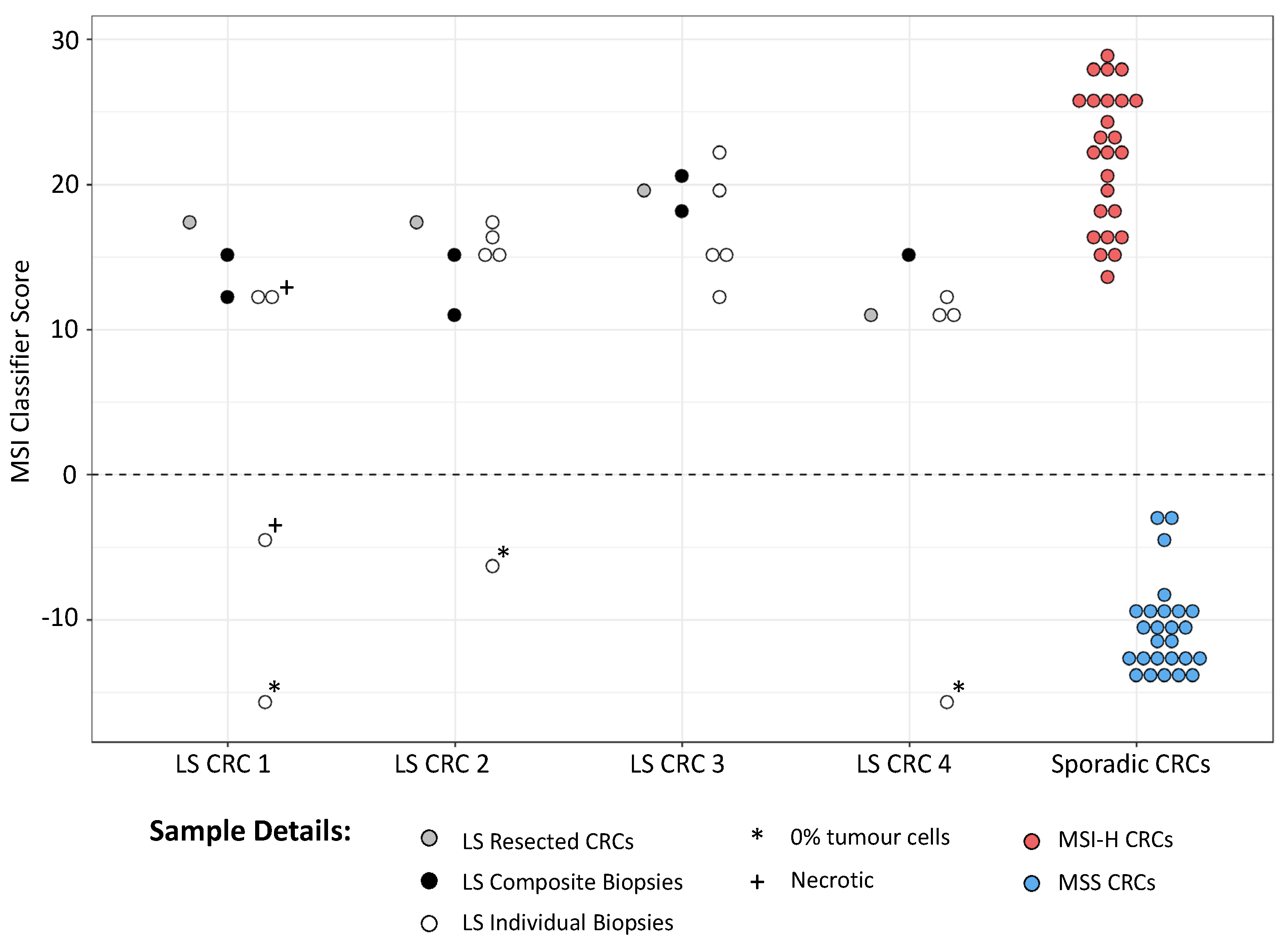
3.2. mPCR Identifies MSI in Individual and Composite CRC Endoscopic Biopsies from LS Patients
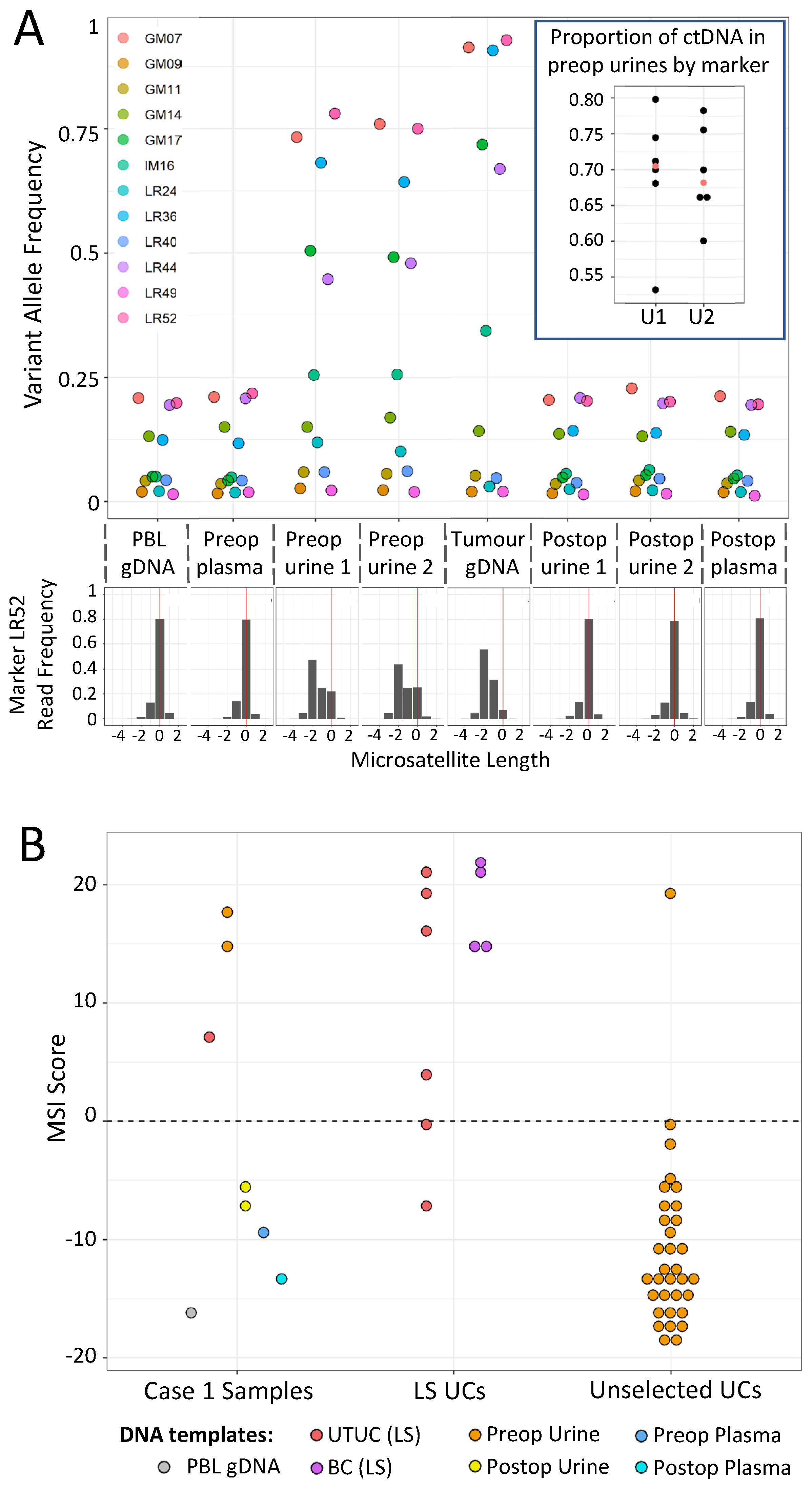
3.3. Case 1: mPCR Can Detect UTUC-Derived MSI-H cfDNA in Urine from an LS Patient
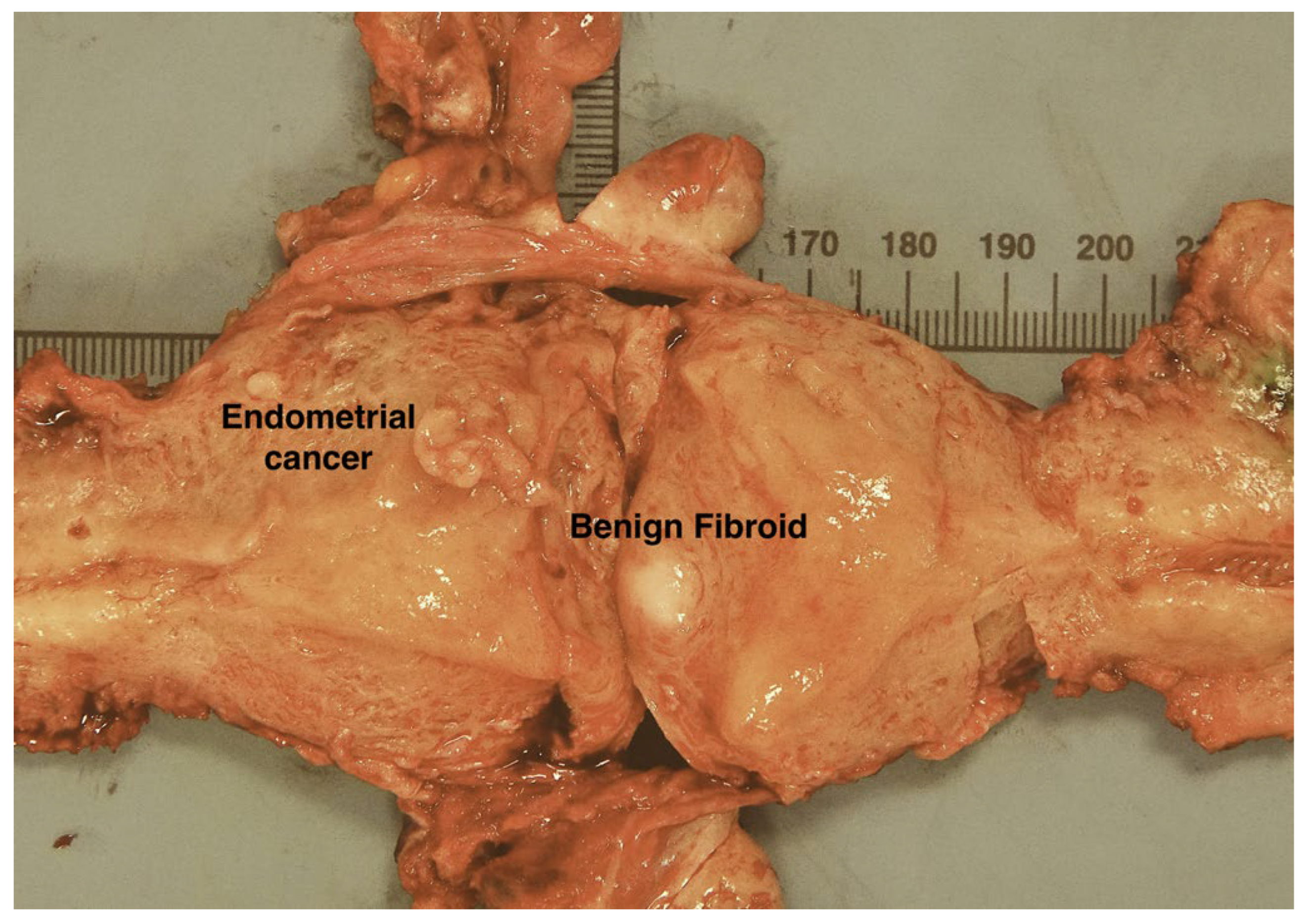
3.4. Case 2: mPCR Can Detect EC-Derived MSI-H cfDNA in Urine from an LS Patient
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lynch, H.T.; Lynch, P.M.; Lanspa, S.J.; Snyder, C.L.; Lynch, J.F.; Boland, C.R. Review of the Lynch syndrome: History, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin. Genet. 2009, 76, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; ten Broeke, S.W.; Plazzer, J.-P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Win, A.K.; Jenkins, M.A.; Dowty, J.G.; Antoniou, A.C.; Lee, A.; Giles, G.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Ahnen, D.J.; et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2017, 26, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, K.; Banno, K.; Yanokura, M.; Kobayashi, Y.; Kisu, I.; Ueki, A.; Ono, A.; Asahara, N.; Nomura, H.; Hirasawa, A.; et al. Relationship between DNA Mismatch Repair Deficiency and Endometrial Cancer. Mol. Biol. Int. 2011, 2011, 256063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelis, G.L.D.; Bottarelli, L.; Azzoni, C.; Angelis, N.D.; Leandro, G.; Di Mario, F.; Gaiani, F.; Negri, F. Microsatellite instability in colorectal cancer. Acta Biomed. 2018, 89, 97–101. [Google Scholar] [CrossRef]
- Järvinen, H.J.; Aarnio, M.; Mustonen, H.; Aktan–Collan, K.; Aaltonen, L.A.; Peltomäki, P.; De La Chapelle, A.; Mecklin, J. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000, 118, 829–834. [Google Scholar] [CrossRef]
- Burn, J.; Sheth, H.; Elliott, F.; Reed, L.; Macrae, F.; Mecklin, J.-P.; Möslein, G.; McRonald, F.E.; Bertario, L.; Evans, D.G.; et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: A double-blind, randomised, placebo-controlled trial. Lancet 2020, 395, 1855–1863. [Google Scholar] [CrossRef]
- Snowsill, T.; Coelho, H.; Huxley, N.; Jones-Hughes, T.; Briscoe, S.; Frayling, I.M.; Hyde, C. Molecular Testing for Lynch Syndrome in People with Colorectal Cancer, Peninsula Technology Assessment Group (PenTAG). University of Exeter Medical School (Report for NICE). Available online: https://www.nice.org.uk/guidance/dg27/documents/diagnostics-assessment-report (accessed on 30 May 2022).
- Crosbie, E.J.; Ryan, N.A.J.; Arends, M.J.; Bosse, T.; Burn, J.; Cornes, J.M.; Crawford, R.; Eccles, D.; Frayling, I.M.; Ghaem-Maghami, S.; et al. The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome. Genet. Med. 2019, 21, 2390–2400. [Google Scholar] [CrossRef] [Green Version]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Balaguer, F.; Sempere, L.; Xicola, R.M.; Bujanda, L.; et al. The efficacy of adjuvant chemotherapy with 5-fluorouracil in colorectal cancer depends on the mismatch repair status. Eur. J. Cancer 2009, 45, 365–373. [Google Scholar] [CrossRef]
- Battaglin, F.; Naseem, M.; Lenz, H.-J.; Salem, M.E. Microsatellite Instability in Colorectal Cancer: Overview of Its Clinical Significance and Novel Perspectives. Clin. Adv. Hematol. Oncol. H&O 2018, 16, 735–745. [Google Scholar]
- Duraturo, F.; Liccardo, R.; De Rosa, M.; Izzo, P. Genetics, diagnosis and treatment of Lynch syndrome: Old lessons and current challenges (Review). Oncol. Lett. 2019, 17, 3048–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Wu, J.; Lv, Y.; Wang, N.; Zhao, Y.; Zhang, P.; Liu, Y.; Chen, A.; Li, J.; Li, X.; Guo, Y.; et al. The value of single-source dual-energy CT imaging for discriminating microsatellite instability from microsatellite stability human colorectal cancer. Eur. Radiol. 2019, 29, 3782–3790. [Google Scholar] [CrossRef]
- Pernicka, J.S.G.; Gagniere, J.; Chakraborty, J.; Yamashita, R.; Nardo, L.; Creasy, J.M.; Petkovska, I.; Do, R.R.K.; Bates, D.D.B.; Paroder, V.; et al. Radiomics-based prediction of microsatellite instability in colorectal cancer at initial computed tomography evaluation. Abdom. Radiol. 2019, 44, 3755–3763. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhong, Q.; Zhang, L.; Wang, M.; Xiao, W.; Cui, F.; Yu, F.; Huang, C.; Feng, Z. Computed Tomography-Based Radiomics Model to Preoperatively Predict Microsatellite Instability Status in Colorectal Cancer: A Multicenter Study. Front. Oncol. 2021, 11, 6786. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, W.; He, D.; Cui, X.; Tian, S.; Yin, H.; Song, B. Development and validation of a radiomics model based on T2WI images for preoperative prediction of microsatellite instability status in rectal cancer. Medicine 2020, 99, e19428. [Google Scholar] [CrossRef]
- Kallenbach-Thieltges, A.; Großerueschkamp, F.; Jütte, H.; Kuepper, C.; Reinacher-Schick, A.; Tannapfel, A.; Gerwert, K. Label-free, automated classification of microsatellite status in colorectal cancer by infrared imaging. Sci. Rep. 2020, 10, 10161. [Google Scholar] [CrossRef]
- Pei, Q.; Yi, X.; Chen, C.; Pang, P.; Fu, Y.; Lei, G.; Chen, C.; Tan, F.; Gong, G.; Li, Q.; et al. Pre-treatment CT-based radiomics nomogram for predicting microsatellite instability status in colorectal cancer. Eur. Radiol. 2021, 32, 714–724. [Google Scholar] [CrossRef]
- Vasen, H.; Abdirahman, M.; Brohet, R.; Langers, A.M.J.; Kleibeuker, J.H.; Van Kouwen, M.; Koornstra, J.J.; Boot, H.; Cats, A.; Dekker, E.; et al. One to 2-Year Surveillance Intervals Reduce Risk of Colorectal Cancer in Families With Lynch Syndrome. Gastroenterology 2010, 138, 2300–2306. [Google Scholar] [CrossRef]
- Seppälä, T.T.; Latchford, A.; Negoi, I.; Soares, A.S.; Jimenez-Rodriguez, R.; Sánchez-Guillén, L.; Evans, D.G.; Ryan, N.; Crosbie, E.J.; Dominguez-Valentin, M.; et al. European guidelines from the EHTG and ESCP for Lynch syndrome: An updated third edition of the Mallorca guidelines based on gene and gender. BJS 2020, 108, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Shia, J.; Stadler, Z.; Weiser, M.R.; Rentz, M.; Gonen, M.; Tang, L.H.; Vakiani, E.; Katabi, N.; Xiong, X.; Markowitz, A.J.; et al. Immunohistochemical Staining for DNA Mismatch Repair Proteins in Intestinal Tract Carcinoma. Am. J. Surg. Pathol. 2011, 35, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Warrier, S.K.; Trainer, A.H.; Lynch, A.C.; Mitchell, C.; Hiscock, R.; Sawyer, S.; Boussioutas, A.; Heriot, A.G. Preoperative Diagnosis of Lynch Syndrome With DNA Mismatch Repair Immunohistochemistry on a Diagnostic Biopsy. Dis. Colon Rectum 2011, 54, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Kumarasinghe, A.P.; de Boer, B.; Bateman, A.C.; Kumarasinghe, M.P. DNA mismatch repair enzyme immunohistochemistry in colorectal cancer: A comparison of biopsy and resection material. Pathology 2010, 42, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Vilkin, A.; Halpern, M.; Morgenstern, S.; Brazovski, E.; Gingold-Belfer, R.; Boltin, D.; Purim, O.; Kundel, Y.; Welinsky, S.; Brenner, B.; et al. How reliable is immunohistochemical staining for DNA mismatch repair proteins performed after neoadjuvant chemoradiation? Hum. Pathol. 2014, 45, 2029–2036. [Google Scholar] [CrossRef]
- Vilkin, A.; Leibovici-Weissman, Y.; Halpern, M.; Morgenstern, S.; Brazovski, E.; Gingold-Belfer, R.; Wasserberg, N.; Brenner, B.; Niv, Y.; Sneh-Arbib, O.; et al. Immunohistochemistry staining for mismatch repair proteins: The endoscopic biopsy material provides useful and coherent results. Hum. Pathol. 2015, 46, 1705–1711. [Google Scholar] [CrossRef]
- O’Brien, O.; Ryan, É.; Creavin, B.; Kelly, M.E.; Mohan, H.M.; Geraghty, R.; Winter, D.C.; Sheahan, K. Correlation of immunohistochemical mismatch repair protein status between colorectal carcinoma endoscopic biopsy and resection specimens. J. Clin. Pathol. 2018, 71, 631–636. [Google Scholar] [CrossRef]
- Gallon, R.; Gawthorpe, P.; Phelps, R.; Hayes, C.; Borthwick, G.; Santibanez-Koref, M.; Jackson, M.; Burn, J. How Should We Test for Lynch Syndrome? A Review of Current Guidelines and Future Strategies. Cancers 2021, 13, 406. [Google Scholar] [CrossRef]
- Gilson, P.; Merlin, J.-L.; Harlé, A. Detection of Microsatellite Instability: State of the Art and Future Applications in Circulating Tumour DNA (ctDNA). Cancers 2021, 13, 1491. [Google Scholar] [CrossRef]
- Gallon, R.; Sheth, H.; Hayes, C.; Redford, L.; Alhilal, G.; O’Brien, O.; Spiewak, H.; Waltham, A.; McAnulty, C.; Izuogu, O.G.; et al. Sequencing-based microsatellite instability testing using as few as six markers for high-throughput clinical diagnostics. Hum. Mutat. 2019, 41, 332–341. [Google Scholar] [CrossRef]
- Gallon, R.; Mühlegger, B.; Wenzel, S.-S.; Sheth, H.; Hayes, C.; Aretz, S.; Dahan, K.; Foulkes, W.; Kratz, C.P.; Ripperger, T.; et al. A sensitive and scalable microsatellite instability assay to diagnose constitutional mismatch repair deficiency by sequencing of peripheral blood leukocytes. Hum. Mutat. 2019, 40, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Batista, R.; Vinagre, J.; Prazeres, H.; Sampaio, C.; Peralta, P.; Conceição, P.; Sismeiro, A.; Leão, R.; Gomes, A.; Furriel, F.; et al. Validation of a Novel, Sensitive, and Specific Urine-Based Test for Recurrence Surveillance of Patients With Non-Muscle-Invasive Bladder Cancer in a Comprehensive Multicenter Study. Front. Genet. 2019, 10, 1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, D.G.; Baxter, L.; Gordon, N.S.; Ott, S.; Savage, R.S.; Beggs, A.; James, J.D.; Lickiss, J.; Green, S.; Wallis, Y.; et al. Multiplex PCR and Next Generation Sequencing for the Non-Invasive Detection of Bladder Cancer. PLoS ONE 2016, 11, e0149756. [Google Scholar] [CrossRef] [PubMed]
- Barrow, P.; Khan, M.; Lalloo, F.; Evans, G.; Hill, J. Systematic review of the impact of registration and screening on colorectal cancer incidence and mortality in familial adenomatous polyposis and Lynch syndrome. Br. J. Surg. 2013, 100, 1719–1731. [Google Scholar] [CrossRef] [PubMed]
- Møller, P. The Prospective Lynch Syndrome Database reports enable evidence-based personal precision health care. Hered. Cancer Clin. Pract. 2020, 18, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.R.; Carter, S.; O’Flynn, H.; Njoku, K.; Barr, C.E.; Narine, N.; Shelton, D.; Rana, D.; Crosbie, E.J. DEveloping Tests for Endometrial Cancer deTection (DETECT): Protocol for a diagnostic accuracy study of urine and vaginal samples for the detection of endometrial cancer by cytology in women with postmenopausal bleeding. BMJ Open 2021, 11, e050755. [Google Scholar] [CrossRef] [PubMed]
- Njoku, K.; Chiasserini, D.; Jones, E.R.; Barr, C.E.; O’Flynn, H.; Whetton, A.D.; Crosbie, E.J. Urinary Biomarkers and Their Potential for the Non-Invasive Detection of Endometrial Cancer. Front. Oncol. 2020, 10, 9016. [Google Scholar] [CrossRef]
- O’Flynn, H.; Ryan, N.A.J.; Narine, N.; Shelton, D.; Rana, D.; Crosbie, E.J. Diagnostic accuracy of cytology for the detection of endometrial cancer in urine and vaginal samples. Nat. Commun. 2021, 12, 952. [Google Scholar] [CrossRef]
- Gervais, A.L.; Marques, M.; Gaudreau, L. PCRTiler: Automated design of tiled and specific PCR primer pairs. Nucleic Acids Res. 2010, 38, W308–W312. [Google Scholar] [CrossRef] [Green Version]
- Holleley, C.E.; Geerts, P.G. Multiplex Manager 1.0: A cross-platform computer program that plans and optimizes multiplex PCR. BioTechniques 2009, 46, 511–517. [Google Scholar] [CrossRef]
- Ståhlberg, A.; Krzyzanowski, P.; Egyud, M.; Filges, S.; Stein, L.; Godfrey, T.E. Simple multiplexed PCR-based barcoding of DNA for ultrasensitive mutation detection by next-generation sequencing. Nat. Protoc. 2017, 12, 664–682. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, S.; Zhan, T.; Betge, J.; Rauscher, B.; Belle, S.; Gutting, T.; Schulte, N.; Jesenofsky, R.; Härtel, N.; Gaiser, T.; et al. Detection of mutational patterns in cell-free DNA of colorectal cancer by custom amplicon sequencing. Mol. Oncol. 2019, 13, 1669–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiatt, J.B.; Pritchard, C.C.; Salipante, S.J.; O’Roak, B.J.; Shendure, J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013, 23, 843–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redford, L.; Alhilal, G.; Needham, S.; O’Brien, O.; Coaker, J.; Tyson, J.; Amorim, L.M.; Middleton, I.; Izuogu, O.; Arends, M.; et al. A novel panel of short mononucleotide repeats linked to informative polymorphisms enabling effective high volume low cost discrimination between mismatch repair deficient and proficient tumours. PLoS ONE 2018, 13, e0203052. [Google Scholar] [CrossRef] [Green Version]
- Piovesan, A.; Pelleri, M.C.; Antonaros, F.; Strippoli, P.; Caracausi, M.; Vitale, L. On the length, weight and GC content of the human genome. BMC Res. Notes 2019, 12, 106. [Google Scholar] [CrossRef]
- Dudley, J.C.; Schroers-Martin, J.; Lazzareschi, D.V.; Shi, W.Y.; Chen, S.B.; Esfahani, M.S.; Trivedi, D.; Chabon, J.J.; Chaudhuri, A.A.; Stehr, H.; et al. Detection and Surveillance of Bladder Cancer Using Urine Tumor DNA. Cancer Discov. 2019, 9, 500–509. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Steward, D.L.; Robinson-Smith, T.M.; Haugen, B.R.; Klopper, J.P.; Zhu, Z.; Fagin, J.A.; Falciglia, M.; Weber, K.; Nikiforova, M.N. Molecular Testing for Mutations in Improving the Fine-Needle Aspiration Diagnosis of Thyroid Nodules. J. Clin. Endocrinol. Metab. 2009, 94, 2092–2098. [Google Scholar] [CrossRef]
- Loukola, A.; Salovaara, R.; Kristo, P.; Moisio, A.-L.; Kääriäinen, H.; Ahtola, H.; Eskelinen, M.; Härkönen, N.; Julkunen, R.; Kangas, E.; et al. Microsatellite Instability in Adenomas as a Marker for Hereditary Nonpolyposis Colorectal Cancer. Am. J. Pathol. 1999, 155, 1849–1853. [Google Scholar] [CrossRef] [Green Version]
- Pino, M.S.; Mino-Kenudson, M.; Wildemore, B.M.; Ganguly, A.; Batten, J.; Sperduti, I.; Iafrate, A.J.; Chung, D.C. Deficient DNA Mismatch Repair Is Common in Lynch Syndrome-Associated Colorectal Adenomas. J. Mol. Diagn. 2009, 11, 238–247. [Google Scholar] [CrossRef] [Green Version]
- Sekine, S.; Mori, T.; Ogawa, R.; Tanaka, M.; Yoshida, H.; Taniguchi, H.; Nakajima, T.; Sugano, K.; Yoshida, T.; Kato, M.; et al. Mismatch repair deficiency commonly precedes adenoma formation in Lynch Syndrome-Associated colorectal tumorigenesis. Mod. Pathol. 2017, 30, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Dabir, P.D.; Bruggeling, C.E.; van der Post, R.S.; Dutilh, B.E.; Hoogerbrugge, N.; Ligtenberg, M.J.L.; Boleij, A.; Nagtegaal, I.D. Microsatellite instability screening in colorectal adenomas to detect Lynch syndrome patients? A systematic review and meta-analysis. Eur. J. Hum. Genet. 2020, 28, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.A.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut 2013, 62, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Fujita, K.; Matsuzaki, K.; Matsushita, M.; Kawamura, N.; Koh, Y.; Nakano, K.; Wang, C.; Ishizuya, Y.; Yamamoto, Y.; et al. Diagnostic potential of TERT promoter and FGFR 3 mutations in urinary cell-free DNA in upper tract urothelial carcinoma. Cancer Sci. 2019, 110, 1771–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiadis, A.; Durham, J.N.; Keefer, L.A.; Bartlett, B.R.; Zielonka, M.; Murphy, D.; White, J.R.; Lu, S.; Verner, E.L.; Ruan, F.; et al. Noninvasive Detection of Microsatellite Instability and High Tumor Mutation Burden in Cancer Patients Treated with PD-1 Blockade. Clin. Cancer Res. 2019, 25, 7024–7034. [Google Scholar] [CrossRef] [Green Version]
- Willis, J.; Lefterova, M.I.; Artyomenko, A.; Kasi, P.M.; Nakamura, Y.; Mody, K.; Catenacci, D.V.; Fakih, M.; Barbacioru, C.; Zhao, J.; et al. Validation of Microsatellite Instability Detection Using a Comprehensive Plasma-Based Genotyping Panel. Clin. Cancer Res. 2019, 25, 7035–7045. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, Z.; Liu, C.; Shi, D.; Li, D.; Zheng, M.; Han-Zhang, H.; Lizaso, A.; Xiang, J.; Lv, J.; et al. Detection of Microsatellite Instability from Circulating Tumor DNA by Targeted Deep Sequencing. J. Mol. Diagn. 2020, 22, 860–870. [Google Scholar] [CrossRef]
- Silveira, A.B.; Bidard, F.-C.; Kasperek, A.; Melaabi, S.; Tanguy, M.-L.; Rodrigues, M.; Bataillon, G.; Cabel, L.; Buecher, B.; Pierga, J.-Y.; et al. High-Accuracy Determination of Microsatellite Instability Compatible with Liquid Biopsies. Clin. Chem. 2020, 66, 606–613. [Google Scholar] [CrossRef]
- Boldrin, E.; Piano, M.; Alfieri, R.; Mazza, M.; Vassallo, L.; Scapinello, A.; Pilati, P.; Curtarello, M. MSI Analysis in Solid and Liquid Biopsies of Gastroesophageal Adenocarcinoma Patients: A Molecular Approach. Int. J. Mol. Sci. 2021, 22, 7244. [Google Scholar] [CrossRef]
- Brisuda, A.; Pazourkova, E.; Soukup, V.; Horinek, A.; Hrbáček, J.; Capoun, O.; Svobodova, I.; Pospisilova, S.; Korabecna, M.; Mares, J.; et al. Urinary Cell-Free DNA Quantification as Non-Invasive Biomarker in Patients with Bladder Cancer. Urol. Int. 2015, 96, 25–31. [Google Scholar] [CrossRef]
- Bartoletti, R.; Cai, T.; Dal Canto, M.; Boddi, V.; Nesi, G.; Piazzini, M. Multiplex Polymerase Chain Reaction for Microsatellite Analysis of Urine Sediment Cells: A Rapid and Inexpensive Method for Diagnosing and Monitoring Superficial Transitional Bladder Cell Carcinoma. J. Urol. 2006, 175, 2032–2037. [Google Scholar] [CrossRef]
- Wild, P.J.; Fuchs, T.; Stoehr, R.; Zimmermann, D.; Frigerio, S.; Padberg, B.; Steiner, I.; Zwarthoff, E.C.; Burger, M.; Denzinger, S.; et al. Detection of Urothelial Bladder Cancer Cells in Voided Urine Can Be Improved by a Combination of Cytology and Standardized Microsatellite Analysis. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1798–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.-F.; Zheng, H.-X.; Li, N.; Yin, Y.; Cheng, C.-X.; Wang, H.-K.; Wu, L.-N.; Xiao, H. Fluorescent Microsatellite Analysis of Urine Sediment in Patients with Urothelial Carcinoma. Urol. Int. 2010, 85, 296–303. [Google Scholar] [CrossRef]
- Bangma, C.H.; Loeb, S.; Busstra, M.; Zhu, X.; El Bouazzaoui, S.; Refos, J.; Van Der Keur, K.A.; Tjin, S.; Franken, C.G.; van Leenders, G.J.; et al. Outcomes of a Bladder Cancer Screening Program Using Home Hematuria Testing and Molecular Markers. Eur. Urol. 2013, 64, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Sciarra, A.; Di Lascio, G.; Del Giudice, F.; Leoncini, P.P.; Salciccia, S.; Gentilucci, A.; Porreca, A.; Chung, B.I.; Di Pierro, G.; Busetto, G.M.; et al. Comparison of the clinical usefulness of different urinary tests for the initial detection of bladder cancer: A systematic review. Curr. Urol. 2021, 15, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Donahue, T.F.; Bagrodia, A.; Audenet, F.; Donoghue, M.T.; Cha, E.K.; Sfakianos, J.P.; Sperling, D.; Al-Ahmadie, H.; Clendenning, M.; Rosty, C.; et al. Genomic Characterization of Upper-Tract Urothelial Carcinoma in Patients With Lynch Syndrome. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Yuan, X.; Liu, T.; Dai, M.; Fan, Y.; Liu, C.; Strååt, K.; Björkholm, M.; Xu, D. Regulatory region mutations of TERT, PLEKHS1 and GPR126 genes as urinary biomarkers in upper tract urothelial carcinomas. J. Cancer 2021, 12, 3853–3861. [Google Scholar] [CrossRef]
- Levine, D.A.; The Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73, Erratum in Nature 2013, 500, 242. [Google Scholar] [CrossRef] [Green Version]
- Ryan, N.A.J.; Walker, T.D.J.; Bolton, J.; ter Haar, N.; Van Wezel, T.; Glaire, M.A.; Church, D.N.; Evans, D.G.; Bosse, T.; Crosbie, E.J. Histological and Somatic Mutational Profiles of Mismatch Repair Deficient Endometrial Tumours of Different Aetiologies. Cancers 2021, 13, 4538. [Google Scholar] [CrossRef]
- Metcalfe, M.J.; Petros, F.G.; Rao, P.; Mork, M.E.; Xiao, L.; Broaddus, R.R.; Matin, S.F. Universal Point of Care Testing for Lynch Syndrome in Patients with Upper Tract Urothelial Carcinoma. J. Urol. 2017, 199, 60–65. [Google Scholar] [CrossRef]
- Urakami, S.; Inoshita, N.; Oka, S.; Miyama, Y.; Nomura, S.; Arai, M.; Sakaguchi, K.; Kurosawa, K.; Okaneya, T. Clinicopathological characteristics of patients with upper urinary tract urothelial cancer with loss of immunohistochemical expression of the DNA mismatch repair proteins in universal screening. Int. J. Urol. 2017, 25, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, N.A.J.; Glaire, M.A.; Blake, D.; Cabrera-Dandy, M.; Evans, D.G.; Crosbie, E.J. The proportion of endometrial cancers associated with Lynch syndrome: A systematic review of the literature and meta-analysis. Genet. Med. 2019, 21, 2167–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phelps, R.; Gallon, R.; Hayes, C.; Glover, E.; Gibson, P.; Edidi, I.; Lee, T.; Mills, S.; Shaw, A.; Heer, R.; et al. Detection of Microsatellite Instability in Colonoscopic Biopsies and Postal Urine Samples from Lynch Syndrome Cancer Patients Using a Multiplex PCR Assay. Cancers 2022, 14, 3838. https://doi.org/10.3390/cancers14153838
Phelps R, Gallon R, Hayes C, Glover E, Gibson P, Edidi I, Lee T, Mills S, Shaw A, Heer R, et al. Detection of Microsatellite Instability in Colonoscopic Biopsies and Postal Urine Samples from Lynch Syndrome Cancer Patients Using a Multiplex PCR Assay. Cancers. 2022; 14(15):3838. https://doi.org/10.3390/cancers14153838
Chicago/Turabian StylePhelps, Rachel, Richard Gallon, Christine Hayes, Eli Glover, Philip Gibson, Ibrahim Edidi, Tom Lee, Sarah Mills, Adam Shaw, Rakesh Heer, and et al. 2022. "Detection of Microsatellite Instability in Colonoscopic Biopsies and Postal Urine Samples from Lynch Syndrome Cancer Patients Using a Multiplex PCR Assay" Cancers 14, no. 15: 3838. https://doi.org/10.3390/cancers14153838
APA StylePhelps, R., Gallon, R., Hayes, C., Glover, E., Gibson, P., Edidi, I., Lee, T., Mills, S., Shaw, A., Heer, R., Ralte, A., McAnulty, C., Santibanez-Koref, M., Burn, J., & Jackson, M. S. (2022). Detection of Microsatellite Instability in Colonoscopic Biopsies and Postal Urine Samples from Lynch Syndrome Cancer Patients Using a Multiplex PCR Assay. Cancers, 14(15), 3838. https://doi.org/10.3390/cancers14153838