
Critical Requirement of SOS1 for Development of BCR/ABL-Driven Chronic Myelogenous Leukemia

 , ,
, , 
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
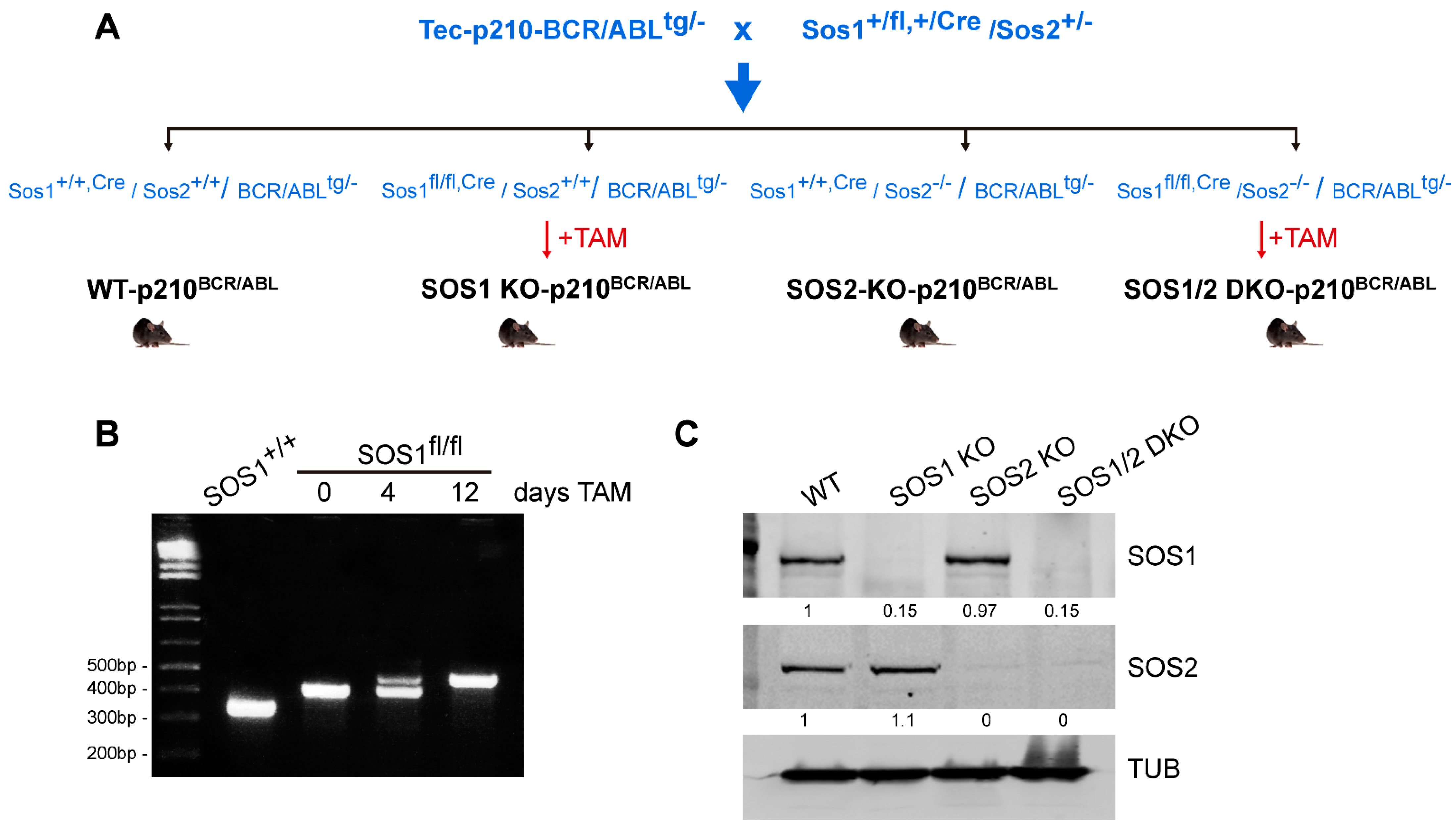
2.1. Mouse Models for CML Analysis
2.2. Peripheral Blood Analysis
2.3. Histological Analyses
2.4. Isolation and Purification of Hematopoietic Stem Progenitor Cells (HSPCs)
2.5. Colony-Forming Unit (CFU) Assays
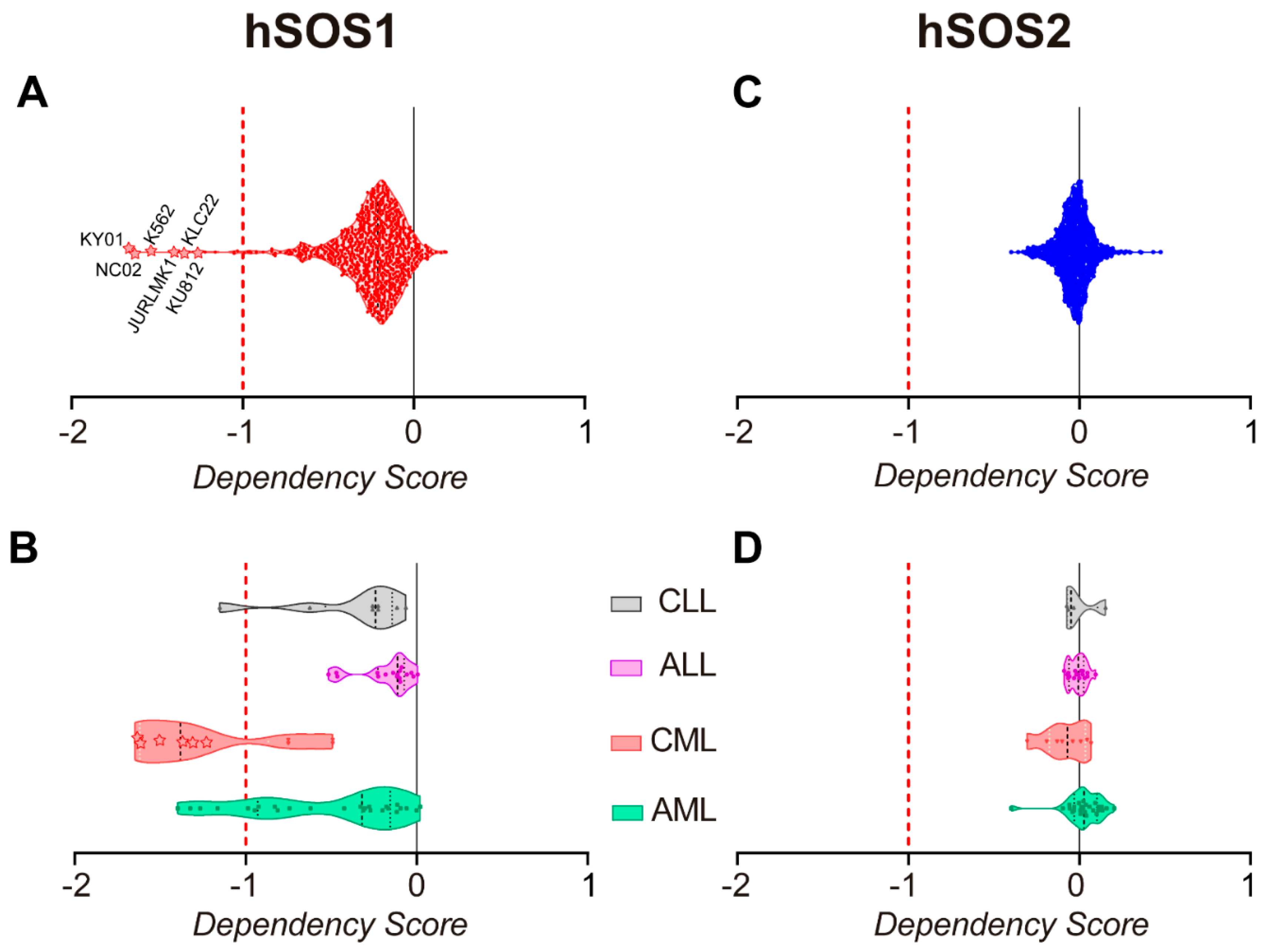
2.6. Pan-Cancer Analysis of Gene Dependency Scores in the DepMap Portal
2.7. Statistical Analysis
3. Results
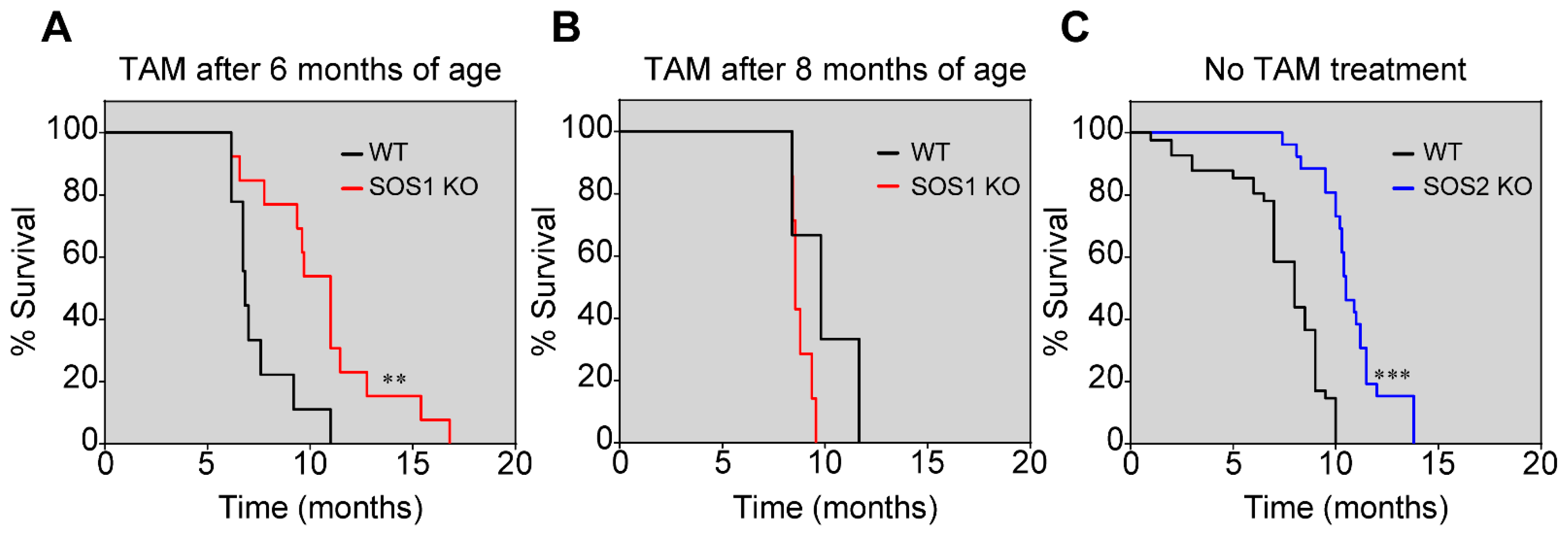
3.1. SOS1 Deficiency Protects from Death in a Murine Model of p210BCR/ABL Induced CML
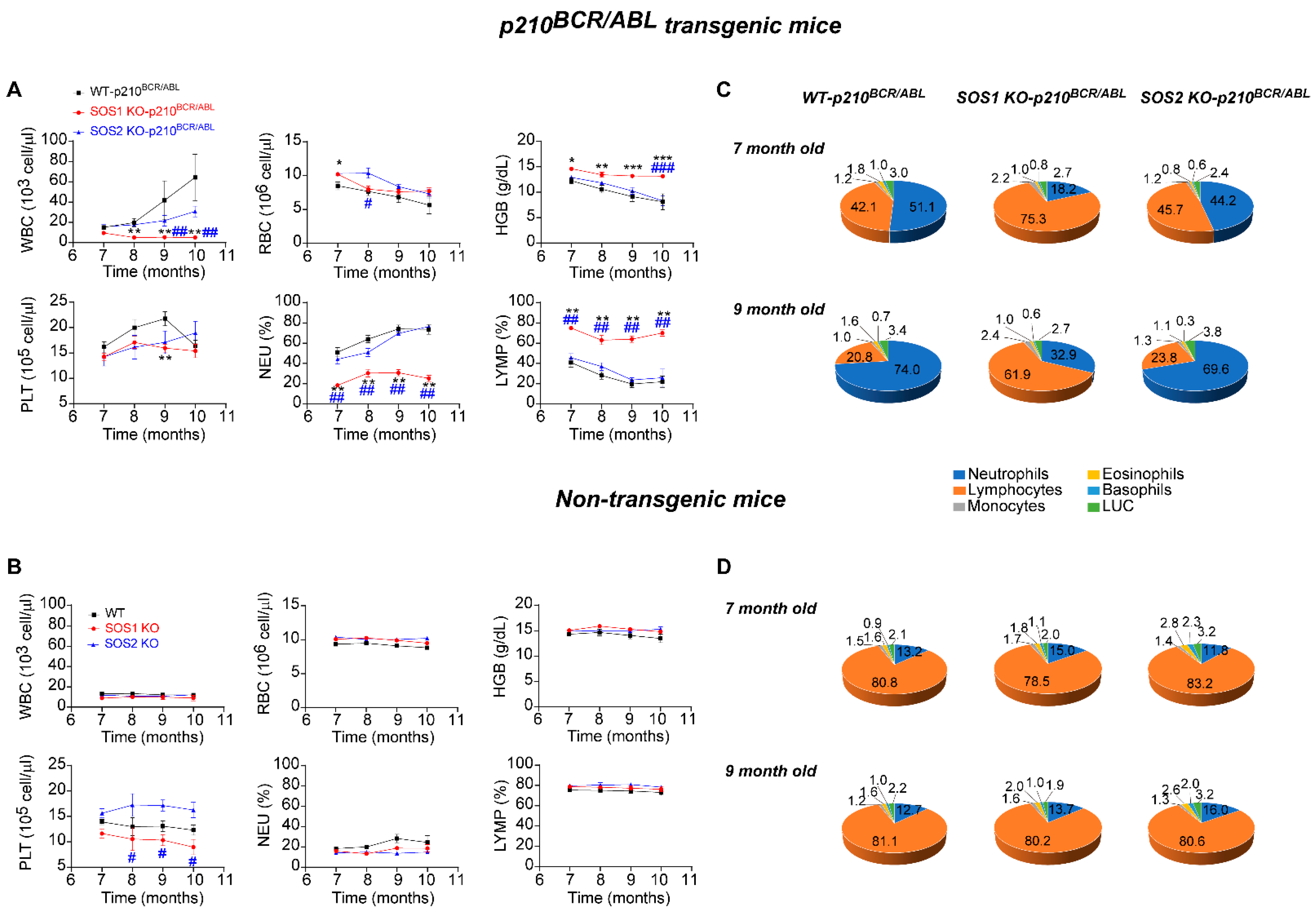
3.2. Reversal of Altered CML Hematological Parameters after SOS1 (But Not SOS2) Ablation in p210BCR/ABL Transgenic Mice
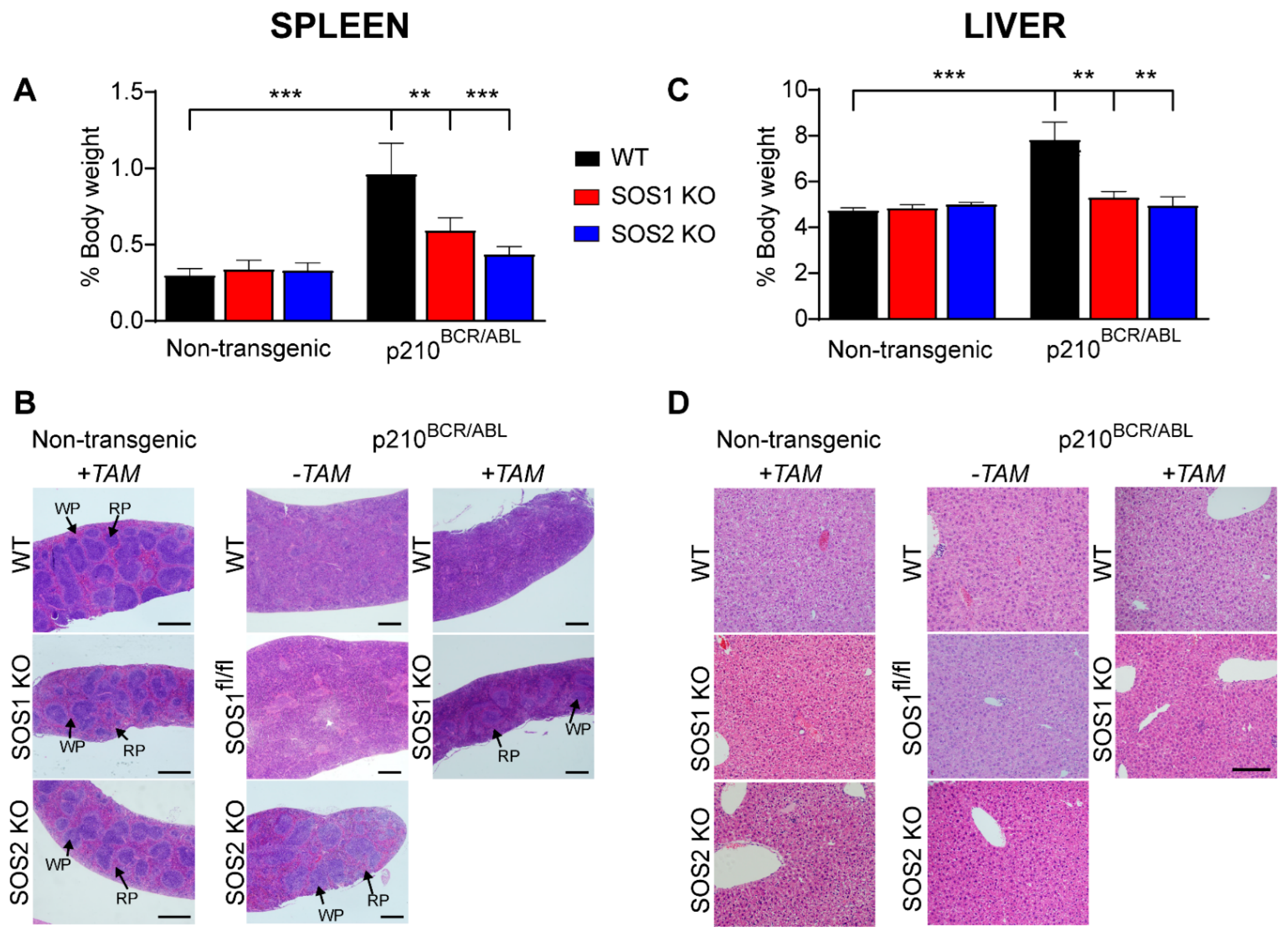
3.3. Loss of SOS1 or SOS2 Reduces Splenomegaly and Hepatomegaly and Restores Spleen Homeostasis in p210BCR/ABL Mice
3.4. Effect of SOS1/2 Genetic Ablation on the Hematopoietic Stem Cell Population of p210BCR/ABL Transgenic Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nowell, P.C.; Hungerford, D.A. Chromosome Studies on Normal and Leukemic Human Leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar] [PubMed]
- Abdulmawjood, B.; Costa, B.; Roma-rodrigues, C.; Baptista, P.V.; Fernandes, A.R. Genetic Biomarkers in Chronic Myeloid Leukemia: What Have We Learned So Far? Int. J. Mol. Sci. 2021, 22, 12516. [Google Scholar] [CrossRef] [PubMed]
- Flis, S.; Chojnacki, T. Chronic Myelogenous Leukemia, a Still Unsolved Problem: Pitfalls and New Therapeutic Possibilities. Drug Des. Dev. Ther. 2019, 13, 825–843. [Google Scholar] [CrossRef] [PubMed]
- Ayatollahi, H.; Keramati, M.R.; Shirdel, A.; Kooshyar, M.M.; Raiszadeh, M.; Shakeri, S.; Sadeghian, M.H. BCR-ABL Fusion Genes and Laboratory Findings in Patients with Chronic Myeloid Leukemia in Northeast Iran. Casp. J. Intern. Med. 2018, 9, 65–70. [Google Scholar] [CrossRef]
- Avelino, K.Y.P.S.; Silva, R.R.; da Silva Junior, A.G.; Oliveira, M.D.L.; Andrade, C.A.S. Smart Applications of Bionanosensors for BCR/ABL Fusion Gene Detection in Leukemia. J. King Saud Univ. Sci. 2017, 29, 413–423. [Google Scholar] [CrossRef]
- Pane, F.; Frigeri, F.; Sindona, M.; Luciano, L.; Ferrara, F.; Cimino, R.; Meloni, G.; Saglio, G.; Salvatore, F.; Rotoli, B. Neutrophilic-Chronic Myeloid Leukemia: A Distinct Disease with a Specific Molecular Marker (BCR/ABL with C3/A2 Junction). Blood 1996, 88, 2410–2414. [Google Scholar] [CrossRef] [PubMed]
- Kaleem, B.; Shahab, S.; Ahmed, N.; Shamsi, T.S. Chronic Myeloid Leukemia-Prognostic Value of Mutations. Asian Pac. J. Cancer Prev. 2015, 16, 7415–7423. [Google Scholar] [CrossRef]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Daley, G.Q.; Mclaughlin, J.; Witte, O.N.; Baltimore, D. The CML-Specific P210 Bcr/Abl Protein, Unlike v-Abl, Does Not Transform NIH/3T3 Fibroblasts. Science 1987, 237, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Kogan, S.C.; Ward, J.M.; Anver, M.R.; Berman, J.J.; Brayton, C.; Cardiff, R.D.; Carter, J.S.; de Coronado, S.; Downing, J.R.; Fredrickson, T.N.; et al. Bethesda Proposals for Classification of Nonlymphoid Hematopoietic Neoplasms in Mice. Blood 2002, 100, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.; Roeder, I.; Seifert, M. Comparative Gene Expression Analysis Reveals Similarities and Differences of Chronic Myeloid Leukemia Phases. Cancers 2022, 14, 256. [Google Scholar] [CrossRef]
- Gutiérrez-Berzal, J.; Castellano, E.; Martín-Encabo, S.; Gutiérrez-Cianca, N.; Hernández, J.M.; Santos, E.; Guerrero, C. Characterization of P87C3G, a Novel, Truncated C3G Isoform That Is Overexpressed in Chronic Myeloid Leukemia and Interacts with Bcr-Abl. Exp. Cell Res. 2006, 312, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.J.; Zhang, B.; Kaartinen, V.; Haataja, L.; Curtis, I.; de Groffen, J.; Heisterkamp, N. Generation of Rac3 Null Mutant Mice: Role of Rac3 in Bcr/Abl-Caused Lymphoblastic Leukemia. Mol. Cell. Biol. 2005, 25, 5777. [Google Scholar] [CrossRef] [PubMed]
- Skorski, T.; Wlodarski, P.; Daheron, L.; Salomoni, P.; Nieborowska-Skorska, M.; Majewski, M.; Wasik, M.; Calabretta, B. BCR/ABL-Mediated Leukemogenesis Requires the Activity of the Small GTP-Binding Protein Rac. Proc. Natl. Acad. Sci. USA 1998, 95, 11858–11862. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.K.; Cancelas, J.A.; Chae, H.D.; Cox, A.D.; Keller, P.J.; Perrotti, D.; Neviani, P.; Druker, B.J.; Setchell, K.D.R.; Zheng, Y.; et al. Rac Guanosine Triphosphatases Represent Integrating Molecular Therapeutic Targets for BCR-ABL-Induced Myeloproliferative Disease. Cancer Cell 2007, 12, 467–478. [Google Scholar] [CrossRef]
- Thomas, E.K.; Cancelas, J.A.; Zheng, Y.; Williams, D.A. Rac GTPases as Key Regulators of P210-BCR-ABL-Dependent Leukemogenesis. Leukemia 2008, 22, 898–904. [Google Scholar] [CrossRef]
- Gerboth, S.; Frittoli, E.; Palamidessi, A.; Baltanas, F.C.; Salek, M.; Rappsilber, J.; Giuliani, C.; Troglio, F.; Rolland, Y.; Pruneri, G.; et al. Phosphorylation of SOS1 on Tyrosine 1196 Promotes Its RAC GEF Activity and Contributes to BCR-ABL Leukemogenesis. Leukemia 2018, 32, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Baltanás, F.C.; Zarich, N.; Rojas-Cabañeros, J.M.; Santos, E. SOS GEFs in Health and Disease. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188445. [Google Scholar] [CrossRef]
- García-Navas, R.; Liceras-Boillos, P.; Gómez, C.; Baltanás, F.C.; Calzada, N.; Nuevo-Tapioles, C.; Cuezva, J.M.; Santos, E. Critical Requirement of SOS1 RAS-GEF Function for Mitochondrial Dynamics, Metabolism, and Redox Homeostasis. Oncogene 2021, 40, 4538–4551. [Google Scholar] [CrossRef] [PubMed]
- Santos, E.; Crespo, P. The RAS-ERK Pathway: A Route for Couples. Sci. Signal. 2018, 11, eaav0917. [Google Scholar] [CrossRef] [PubMed]
- Nebreda, A.R.; Porras, A.; Santos, E. P21ras-Induced Meiotic Maturation of Xenopus Oocytes in the Absence of Protein Synthesis: MPF Activation Is Preceded by Activation of MAP and S6 Kinases. Oncogene 1993, 8, 467–477. [Google Scholar] [PubMed]
- Guerrero, C.; Martín-Encabo, S.; Fernández-Medarde, A.; Santos, E. C3G-Mediated Suppression of Oncogene-Induced Focus Formation in Fibroblasts Involves Inhibition of ERK Activation, Cyclin A Expression and Alterations of Anchorage-Independent Growth. Oncogene 2004, 23, 4885–4893. [Google Scholar] [CrossRef]
- Fernández-Medarde, A.; Porteros, A.; de las Rivas, J.; Núñez, A.; Fuster, J.J.; Santos, E. Laser Microdissection and Microarray Analysis of the Hippocampus of Ras-GRF1 Knockout Mice Reveals Gene Expression Changes Affecting Signal Transduction Pathways Related to Memory and Learning. Neuroscience 2007, 146, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Esteban, L.; Vass, W.C.; Upadhyaya, C.; Papageorge, A.G.; Yienger, K.; Ward, J.M.; Lowy, D.R.; Santos, E. The Sos1 and Sos2 Ras-Specific Exchange Factors: Differences in Placental Expression and Signaling Properties. EMBO J. 2000, 19, 642–654. [Google Scholar] [CrossRef]
- Esteban, L.M.; Fernández-Medarde, A.; López, E.; Yienger, K.; Guerrero, C.; Ward, J.M.; Tessarollo, L.; Santos, E. Ras-Guanine Nucleotide Exchange Factor Sos2 Is Dispensable for Mouse Growth and Development. Mol. Cell. Biol. 2000, 20, 6410–6413. [Google Scholar] [CrossRef] [PubMed]
- Liceras-Boillos, P.; García-Navas, R.; Ginel-Picardo, A.; Anta, B.; Pérez-Andrés, M.; Lillo, C.; Gómez, C.; Jimeno, D.; Fernández-Medarde, A.; Baltanás, F.C.; et al. Sos1 Disruption Impairs Cellular Proliferation and Viability through an Increase in Mitochondrial Oxidative Stress in Primary MEFs. Oncogene 2016, 35, 6389–6402. [Google Scholar] [CrossRef] [PubMed]
- Baltanás, F.C.; Mucientes-valdivieso, C.; Francisco Lorenzo-Martín, L.; Fernández-parejo, N.; García-navas, R.; Segrelles, C.; Calzada, N.; Fuentes-mateos, R.; Paramio, J.M.; Bustelo, X.R.; et al. Functional Specificity of the Members of the Sos Family of Ras-gef Activators: Novel Role of Sos2 in Control of Epidermal Stem Cell Homeostasis. Cancers 2021, 13, 2152. [Google Scholar] [CrossRef]
- Fernández-Medarde, A.; Fuentes-Mateos, R.; García-Navas, R.; Juan, A.O.S.; Sánchez-López, J.M.; Fernández-Medarde, A.; Santos, E. Anthraquinones as Inhibitors of SOS RAS-GEF Activity. Biomolecules 2021, 11, 1128. [Google Scholar] [CrossRef]
- Zarich, N.; Anta, B.; Fernández-Medarde, A.; Ballester, A.; de Lucas, M.P.; Cámara, A.B.; Anta, B.; Oliva, J.L.; Rojas-Cabañeros, J.M.; Santos, E. The CSN3 Subunit of the COP9 Signalosome Interacts with the HD Region of Sos1 Regulating Stability of This GEF Protein. Oncogenesis 2019, 8, 2. [Google Scholar] [CrossRef]
- Suire, S.; Baltanas, F.C.; Segonds-Pichon, A.; Davidson, K.; Santos, E.; Hawkins, P.T.; Stephens, L.R. Frontline Science: TNF-α and GM-CSF1 Priming Augments the Role of SOS1/2 in Driving Activation of Ras, PI3K-γ, and Neutrophil Proinflammatory Responses. J. Leukoc. Biol. 2019, 106, 815–822. [Google Scholar] [CrossRef]
- Liceras-Boillos, P.; Jimeno, D.; García-Navas, R.; Lorenzo-Martín, L.F.; Menacho-Marquez, M.; Segrelles, C.; Gómez, C.; Calzada, N.; Fuentes-Mateos, R.; Paramio, J.M.; et al. Differential Role of the RasGEFs Sos1 and Sos2 in Mouse Skin Homeostasis and Carcinogenesis. Mol. Cell. Biol. 2018, 38, e00049-18. [Google Scholar] [CrossRef]
- Sini, P.; Cannas, A.; Koleske, A.J.; di Fiore, P.P.; Scita, G. Abl-Dependent Tyrosine Phosphorylation of Sos-1 Mediates Growth-Factor-Induced Rac Activation. Nat. Cell Biol. 2004, 6, 268–274. [Google Scholar] [CrossRef]
- Honda, H.; Oda, H.; Suzuki, T.; Takahashi, T.; Witte, O.N.; Ozawa, K.; Ishikawa, T.; Yazaki, Y.; Hirai, H. Development of Acute Lymphoblastic Leukemia and Myeloproliferative Disorder in Transgenic Mice Expressing P210bcr/Abl: A Novel Transgenic Model for Human Ph1-Positive Leukemias. Blood 1998, 91, 2067–2075. [Google Scholar] [CrossRef]
- Honda, H.; Hirai, H. Model Mice for BCR/ABL-Positive Leukemias. Blood Cells Mol. Dis. 2001, 27, 265–278. [Google Scholar] [CrossRef]
- Baltanás, F.C.; Pérez-Andrés, M.; Ginel-Picardo, A.; Diaz, D.; Jimeno, D.; Liceras-Boillos, P.; Kortum, R.L.; Samelson, L.E.; Orfao, A.; Santos, E. Functional Redundancy of Sos1 and Sos2 for Lymphopoiesis and Organismal Homeostasis and Survival. Mol. Cell Biol. 2013, 33, 4562–4578. [Google Scholar] [CrossRef]
- Muroi, K.; Nakamurat, M.; Amemiya, Y.; Suda, T.; Miura, Y. Expression of C-Kit Receptor (CD 117) and CD34 in Leukemic Cells. Leuk. Lymphoma 2009, 16, 297–305. [Google Scholar] [CrossRef]
- Dybko, J.; Haus, O.; Jaźwiec, B.; Urbaniak, J.; Woźniak, M.; Kaczmar-Dybko, A.; Urbaniak-Kujda, D.; Kapelko-Slowik, K.; Kuliczkowski, K. CD117 (c-Kit) Expression on CD34+ Cells Participates in the Cytogenetic Response to Imatinib in Patients with Chronic Myeloid Leukemia in the First Chronic Phase. Acta Haematol. 2014, 132, 166–171. [Google Scholar] [CrossRef]
- Redondo Monte, E.; Leubolt, G.; Windisch, R.; Kerbs, P.; Dutta, S.; Landspersky, T.; Istvánffy, R.; Oostendorp, R.A.J.; Chen-Wichmann, L.; Herold, T.; et al. Specific Effects of Somatic GATA2 Zinc Finger Mutations on Erythroid Differentiation. Exp. Hematol. 2022, 108, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Kesarwani, M.; Kincaid, Z.; Gomaa, A.; Huber, E.; Rohrabaugh, S.; Siddiqui, Z.; Bouso, M.F.; Latif, T.; Xu, M.; Komurov, K.; et al. Targeting C-FOS and DUSP1 Abrogates Intrinsic Resistance to Tyrosine-Kinase Inhibitor Therapy in BCR-ABL-Induced Leukemia. Nat. Med. 2017, 23, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [PubMed]
- Schemionek, M.; Spieker, T.; Kerstiens, L.; Elling, C.; Essers, M.; Trumpp, A.; Berdel, W.E.; Müller-Tidow, C.; Koschmieder, S. Leukemic Spleen Cells Are More Potent than Bone Marrow-Derived Cells in a Transgenic Mouse Model of CML. Leukemia 2012, 26, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.; Pathak, R.; Martin, M.G.; Bhatt, V.R. Characteristics and Survival of BCR/ABL Negative Chronic Myeloid Leukemia: A Retrospective Analysis of the Surveillance, Epidemiology and End Results Database. Ther. Adv. Hematol. 2015, 6, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Mojtahedi, H.; Yazdanpanah, N.; Rezaei, N. Chronic Myeloid Leukemia Stem Cells: Targeting Therapeutic Implications. Stem Cell Res. Ther. 2021, 12, 603. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; de Santis, S.; Monaldi, C.; Bruno, S.; Mancini, M. Targeting Leukemic Stem Cells in Chronic Myeloid Leukemia: Is It Worth the Effort? Int. J. Mol. Sci. 2021, 22, 7093. [Google Scholar] [CrossRef]
- Faber, Z.J.; Chen, X.; Gedman, A.L.; Boggs, K.; Cheng, J.; Ma, J.; Radtke, I.; Chao, J.R.; Walsh, M.P.; Song, G.; et al. The Genomic Landscape of Core-Binding Factor Acute Myeloid Leukemias. Nat. Genet. 2016, 48, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Jeng, H.H.; Taylor, L.J.; Bar-Sagi, D. Sos-Mediated Cross-Activation of Wild-Type Ras by Oncogenic Ras Is Essential for Tumorigenesis. Nat. Commun. 2012, 3, 1168. [Google Scholar] [CrossRef]
- You, X.; Kong, G.; Ranheim, E.A.; Yang, D.; Zhou, Y.; Zhang, J. Unique Dependence on Sos1 in Kras G12D -Induced Leukemogenesis. Blood 2018, 132, 2575–2579. [Google Scholar] [CrossRef] [PubMed]
- Dempster, J.M.; Rossen, J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2019. [Google Scholar] [CrossRef]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A CRISPR Cell Population Dynamics Model. bioRxiv 2021. [Google Scholar] [CrossRef]
- Pacini, C.; Dempster, J.M.; Boyle, I.; Gonçalves, E.; Najgebauer, H.; Karakoc, E.; van der Meer, D.; Barthorpe, A.; Lightfoot, H.; Jaaks, P.; et al. Integrated Cross-Study Datasets of Genetic Dependencies in Cancer. Nat. Commun. 2021, 12, 1661. [Google Scholar] [CrossRef] [PubMed]
- Panina, S.B.; Baran, N.; Brasil da Costa, F.H.; Konopleva, M.; Kirienko, N.V. A Mechanism for Increased Sensitivity of Acute Myeloid Leukemia to Mitotoxic Drugs. Cell Death Dis. 2019, 10, 617. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting Mitochondrial Oxidative Phosphorylation Eradicates Therapy-Resistant Chronic Myeloid Leukemia Stem Cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Su, R.; Yin, Z.; Huang, G.; Yang, J.; Li, Z.; Zhang, K.; Fei, J. Targeting SOS1 Overcomes Imatinib Resistance with BCR-ABL Independence through Uptake Transporter SLC22A4 in CML. Mol. Ther Oncolytics 2021, 23, 560–570. [Google Scholar] [CrossRef] [PubMed]
- de Beauchamp, L.; Himonas, E.; Helgason, G.V. Mitochondrial Metabolism as a Potential Therapeutic Target in Myeloid Leukaemia. Leukemia 2021, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The Clinical KRAS (G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J.; et al. Discovery of Potent SOS1 Inhibitors That Block RAS Activation via Disruption of the RAS–SOS1 Interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, D.; Gmachl, M.; Ramharter, J.; Teh, J.; Fu, S.-C.; Trapani, F.; Kessler, D.; Rumpel, K.; Botesteanu, D.-A.; Ettmayer, P.; et al. Abstract 1091: BI-3406 and BI 1701963: Potent and Selective SOS1::KRAS Inhibitors Induce Regressions in Combination with MEK Inhibitors or Irinotecan. Cancer Res. 2020, 80, 1091. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.H.; Gerlach, D.; Misale, S.; Petronczki, M.; Kraut, N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov. 2022, 12, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Kumar, V.; Gupta, S.K.; Kumari, G.; Verma, M. Combating TKI Resistance in CML by Inhibiting the PI3K/Akt/MTOR Pathway in Combination with TKIs: A Review. Med. Oncol. 2021, 38, 10. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez, C.; Garcia-Navas, R.; Baltanás, F.C.; Fuentes-Mateos, R.; Fernández-Medarde, A.; Calzada, N.; Santos, E. Critical Requirement of SOS1 for Development of BCR/ABL-Driven Chronic Myelogenous Leukemia. Cancers 2022, 14, 3893. https://doi.org/10.3390/cancers14163893
Gómez C, Garcia-Navas R, Baltanás FC, Fuentes-Mateos R, Fernández-Medarde A, Calzada N, Santos E. Critical Requirement of SOS1 for Development of BCR/ABL-Driven Chronic Myelogenous Leukemia. Cancers. 2022; 14(16):3893. https://doi.org/10.3390/cancers14163893
Chicago/Turabian StyleGómez, Carmela, Rósula Garcia-Navas, Fernando C. Baltanás, Rocío Fuentes-Mateos, Alberto Fernández-Medarde, Nuria Calzada, and Eugenio Santos. 2022. "Critical Requirement of SOS1 for Development of BCR/ABL-Driven Chronic Myelogenous Leukemia" Cancers 14, no. 16: 3893. https://doi.org/10.3390/cancers14163893
APA StyleGómez, C., Garcia-Navas, R., Baltanás, F. C., Fuentes-Mateos, R., Fernández-Medarde, A., Calzada, N., & Santos, E. (2022). Critical Requirement of SOS1 for Development of BCR/ABL-Driven Chronic Myelogenous Leukemia. Cancers, 14(16), 3893. https://doi.org/10.3390/cancers14163893