
Dissecting Molecular Heterogeneity of Circulating Tumor Cells (CTCs) from Metastatic Breast Cancer Patients through Copy Number Aberration (CNA) and Single Nucleotide Variant (SNV) Single Cell Analysis

 ,
,  , ,
, ,  , ,
, ,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. CTC Enrichment, Recovery and Whole Genome Amplification (WGA)
2.3. Library Preparation and Sequencing
2.4. Bioinformatic and Statistical Analyses
3. Results
3.1. CTC Isolation in MBC Patients and Quality Assessment
3.2. Single Nucleotide Variant (SNV) Analysis on Single CTCs
3.3. Single Cell Profiling of CTCs
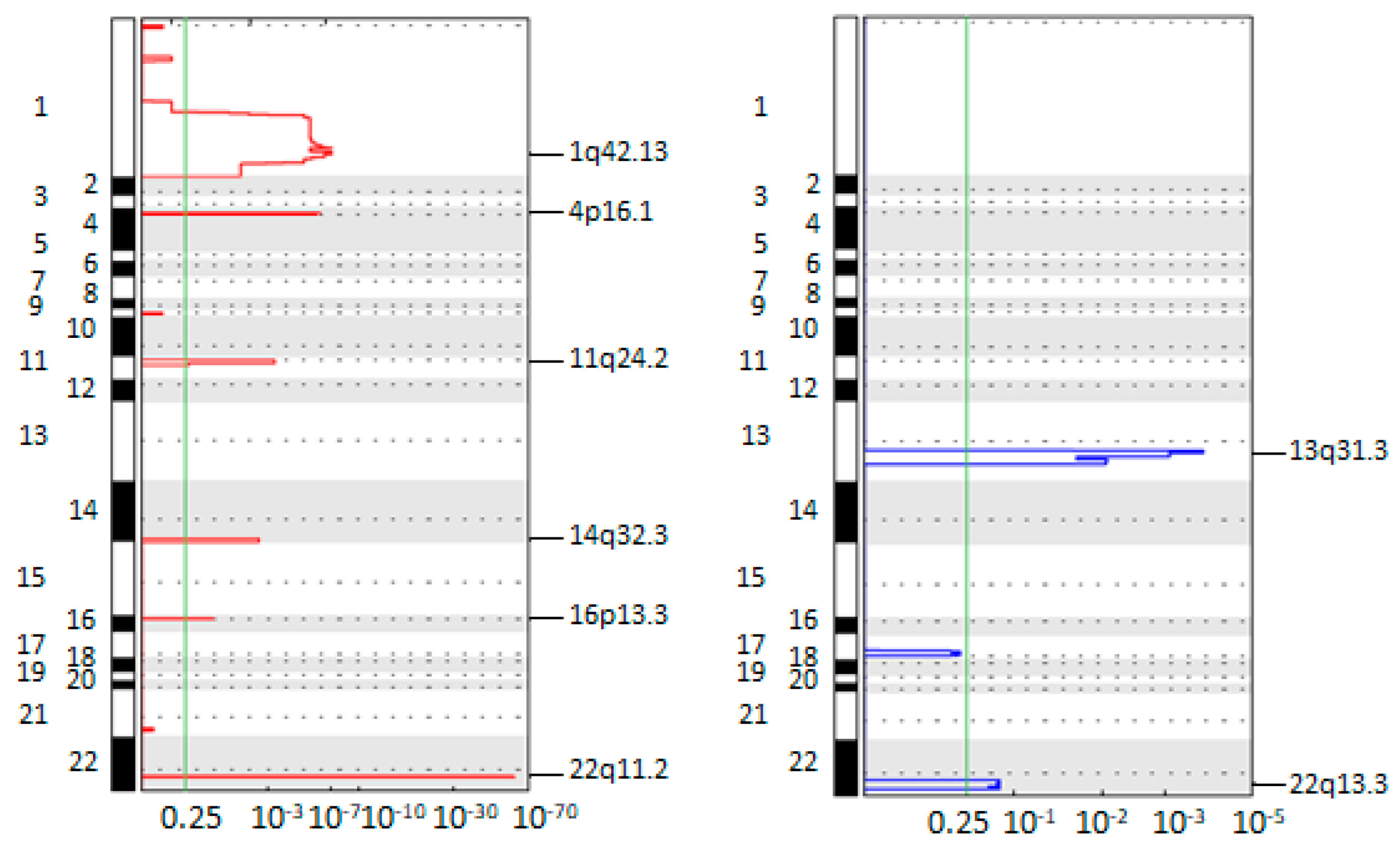
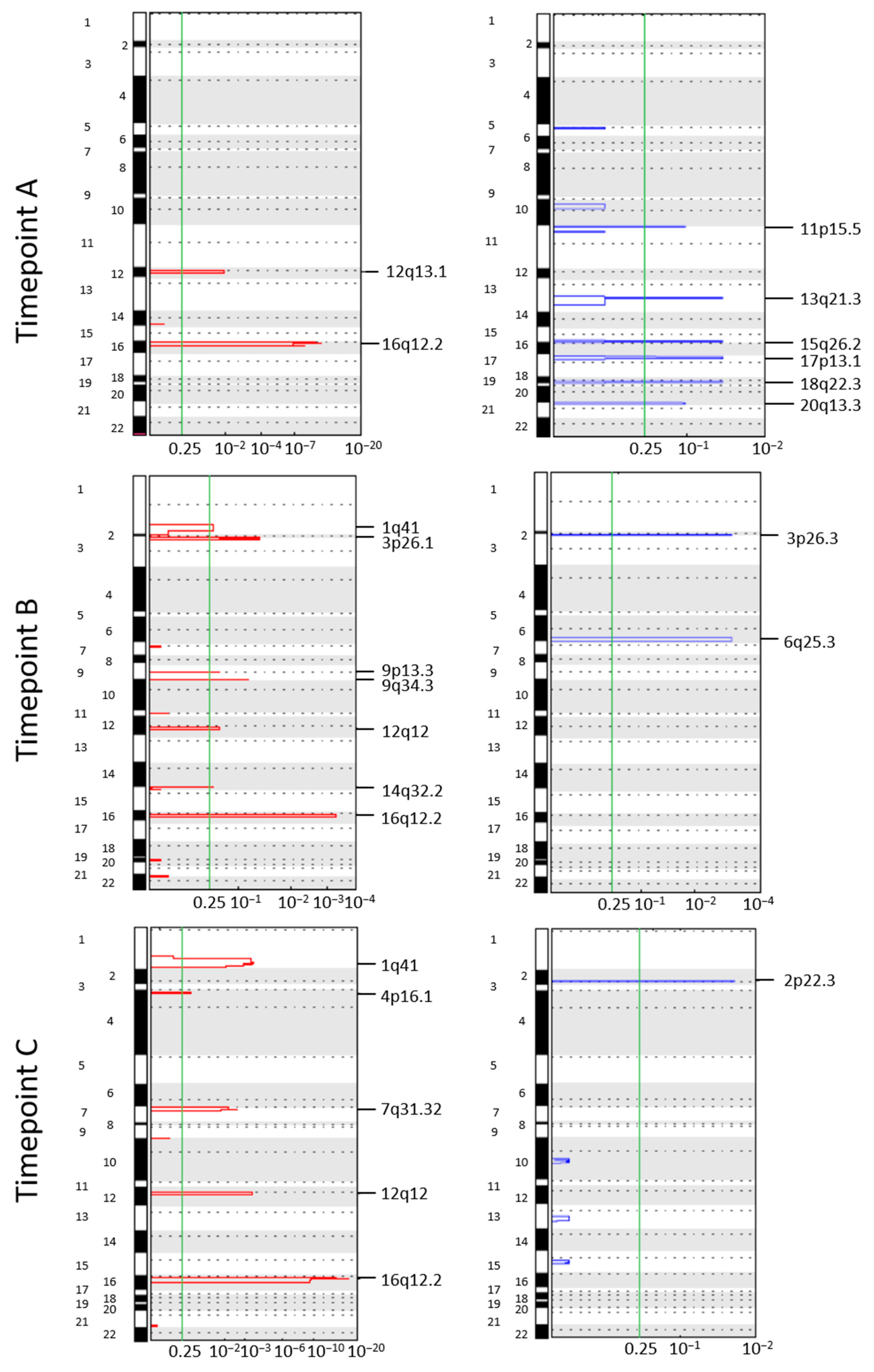
3.4. Longitudinal Investigation of CTC CNA Profiles
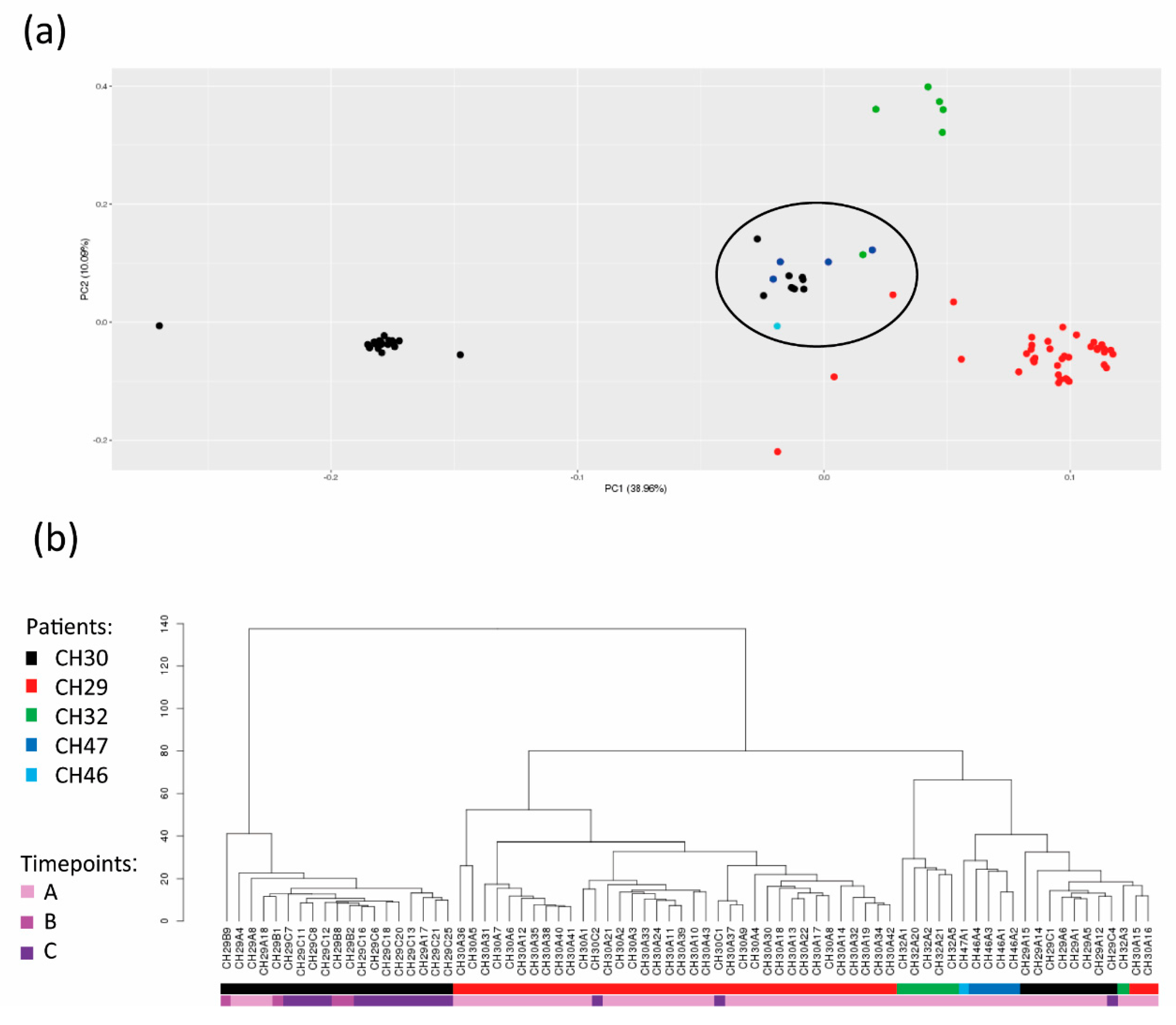
3.5. Identification of a Population of CTCs with Common Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Liu, X.; Liao, X.; He, J.; Niu, L. The Clinicopathological Features and Survival Outcomes of Patients with Different Metastatic Sites in Stage IV Breast Cancer. BMC Cancer 2019, 19, 1091. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Circulating Tumor Cells: Liquid Biopsy of Cancer. Clin. Chem. 2013, 59, 110–118. [Google Scholar] [CrossRef]
- Giordano, A.; Cristofanilli, M. CTCs in Metastatic Breast Cancer. Recent Results Cancer Res. 2012, 195, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, M.; Giordano, A.; Jackson, S.; De Giorgi, U.; Mego, M.; Cohen, E.N.; Gao, H.; Anfossi, S.; Handy, B.C.; Ueno, N.T.; et al. Circulating Tumor Cells as Early Predictors of Metastatic Spread in Breast Cancer Patients with Limited Metastatic Dissemination. Breast Cancer Res. 2014, 16, 440. [Google Scholar] [CrossRef]
- Rossi, T.; Gallerani, G.; Angeli, D.; Cocchi, C.; Bandini, E.; Fici, P.; Gaudio, M.; Martinelli, G.; Rocca, A.; Maltoni, R.; et al. Single-Cell NGS-Based Analysis of Copy Number Alterations Reveals New Insights in Circulating Tumor Cells Persistence in Early-Stage Breast Cancer. Cancers 2020, 12, 2490. [Google Scholar] [CrossRef]
- Bruno, S.; Bochicchio, M.T.; Franchini, E.; Padella, A.; Marconi, G.; Ghelli Luserna di Rorà, A.; Venturi, C.; Raffini, M.; Prisinzano, G.; Ferrari, A.; et al. Identification of Two DNMT3A Mutations Compromising Protein Stability and Methylation Capacity in Acute Myeloid Leukemia. J. Oncol. 2019, 2019, 5985923. [Google Scholar] [CrossRef]
- Sturgill, E.G.; Misch, A.; Lachs, R.; Jones, C.C.; Schlauch, D.; Jones, S.F.; Shastry, M.; Yardley, D.A.; Burris, H.A.; Spigel, D.R.; et al. Next-Generation Sequencing of Patients with Breast Cancer in Community Oncology Clinics. JCO Precis. Oncol. 2021, 5, 1297–1311. [Google Scholar] [CrossRef]
- Goodman, A.M.; Choi, M.; Wieduwilt, M.; Mulroney, C.; Costello, C.; Frampton, G.; Miller, V.; Kurzrock, R. Next-Generation Sequencing Reveals Potentially Actionable Alterations in the Majority of Patients with Lymphoid Malignancies. JCO Precis. Oncol. 2017, 1, 1–13. [Google Scholar] [CrossRef]
- Paoletti, C.; Cani, A.K.; Larios, J.M.; Hovelson, D.H.; Aung, K.; Darga, E.P.; Cannell, E.M.; Baratta, P.J.; Liu, C.J.; Chu, D.; et al. Comprehensive Mutation and Copy Number Profiling in Archived Circulating Breast Cancer Tumor Cells Documents Heterogeneous Resistance Mechanisms. Cancer Res. 2018, 78, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A Tool for Assessing Copy Number and Allelic Content Using next-Generation Sequencing Data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Möhlendick, B.; Bartenhagen, C.; Behrens, B.; Honisch, E.; Raba, K.; Knoefel, W.T.; Stoecklein, N.H. A Robust Method to Analyze Copy Number Alterations of Less than 100 Kb in Single Cells Using Oligonucleotide Array CGH. PLoS ONE 2013, 8, e67031. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 Facilitates Sensitive and Confident Localization of the Targets of Focal Somatic Copy-Number Alteration in Human Cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, R.; Spring, L.; O’Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 Mutations and Acquired Resistance to CDK 4/6 Inhibitors in Patients with Metastatic Breast Cancer. Ann. Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Gymnopoulos, M.; Elsliger, M.-A.; Vogt, P.K. Rare Cancer-Specific Mutations in PIK3CA Show Gain of Function. Proc. Natl. Acad. Sci. USA 2007, 104, 5569–5574. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F.; Rotunno, G.; Salvianti, F.; Galardi, F.; Pestrin, M.; Gabellini, S.; Simi, L.; Mancini, I.; Vannucchi, A.M.; Pazzagli, M.; et al. Mutational Analysis of Single Circulating Tumor Cells by next Generation Sequencing in Metastatic Breast Cancer. Oncotarget 2016, 7, 26107–26119. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.P.; Kiliti, A.J.; Zhang, K.; Vasani, N.; Mao, N.; Jordan, E.; Wise, H.C.; Shrestha Bhattarai, T.; Hu, W.; Dorso, M.; et al. AKT1 E17K Inhibits Cancer Cell Migration by Abrogating β-Catenin Signaling. Mol. Cancer Res. 2021, 19, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.; Anzeneder, T.; Schulz, A.; Beckmann, G.; Byrne, A.T.; Jeffers, M.; Pena, C.; Politz, O.; Köchert, K.; Vonk, R.; et al. AKT1 E17K Mutation Profiling in Breast Cancer: Prevalence, Concurrent Oncogenic Alterations, and Blood-Based Detection. BMC Cancer 2016, 16, 622. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, L.; Dong, Y.; Tao, C.; Zhang, R.; Shao, H.; Shen, H. Effect of AKT1 (p. E17K) Hotspot Mutation on Malignant Tumorigenesis and Prognosis. Front. Cell Dev. Biol. 2020, 8, 573599. [Google Scholar] [CrossRef]
- Fernández-Madrid, F.; Tang, N.; Alansari, H.; Granda, J.L.; Tait, L.; Amirikia, K.C.; Moroianu, M.; Wang, X.; Karvonen, R.L. Autoantibodies to Annexin XI-A and Other Autoantigens in the Diagnosis of Breast Cancer. Cancer Res. 2004, 64, 5089–5096. [Google Scholar] [CrossRef]
- Elsheikh, S.; Green, A.R.; Aleskandarany, M.A.; Grainge, M.; Paish, C.E.; Lambros, M.B.K.; Reis-Filho, J.S.; Ellis, I.O. CCND1 Amplification and Cyclin D1 Expression in Breast Cancer and Their Relation with Proteomic Subgroups and Patient Outcome. Breast Cancer Res. Treat. 2008, 109, 325–335. [Google Scholar] [CrossRef]
- Castells, A.; Gusella, J.F.; Ramesh, V.; Rustgi, A.K. A Region of Deletion on Chromosome 22q13 Is Common to Human Breast and Colorectal Cancers. Cancer Res. 2000, 60, 2836–2839. [Google Scholar]
- Li, Z.; Zou, W.; Zhang, J.; Zhang, Y.; Xu, Q.; Li, S.; Chen, C. Mechanisms of CDK4/6 Inhibitor Resistance in Luminal Breast Cancer. Front. Pharmacol. 2020, 11, 580251. [Google Scholar] [CrossRef]
- Karlseder, J.; Zeillinger, R.; Schneeberger, C.; Czerwenka, K.; Speiser, P.; Kubista, E.; Birnbaum, D.; Gaudray, P.; Theillet, C. Patterns of Dna Amplification at Band Q13 of Chromosome 11 in Human Breast Cancer. Genes Chromosom. Cancer 1994, 9, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Ormandy, C.J.; Musgrove, E.A.; Hui, R.; Daly, R.J.; Sutherland, R.L. Cyclin D1, EMS1 and 11q13 Amplification in Breast Cancer. Breast Cancer Res. Treat. 2003, 78, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Rhiem, K.; Klein, A.; Münch, M.; Kreutzfeld, R.; Ramser, J.; Wardelmann, E.; Schackert, G.; von Deimling, A.; Wiestler, O.D.; Schmutzler, R.K. Chromosomal Region 15q21.1 Is a Frequent Target of Allelic Imbalance in Advanced Breast Carcinomas. Int. J. Cancer 2003, 106, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Petersen, I.; Schmutzler, R.K.; Wolfarth, B.; Lenartz, D.; Bierhoff, E.; Hümmerich, J.; Müller, D.J.; Stangl, A.P.; Schramm, J.; et al. Evidence for a Novel Tumor Suppressor Gene on Chromosome 15 Associated with Progression to a Metastatic Stage in Breast Cancer. Oncogene 1996, 12, 973–978. [Google Scholar] [PubMed]
- Poetsch, M.; Kleist, B. Loss of Heterozygosity at 15q21.3 Correlates with Occurrence of Metastases in Head and Neck Cancer. Mod. Pathol. 2006, 19, 1462–1469. [Google Scholar] [CrossRef]
- Kee, H.J.; Shin, J.H.; Chang, J.; Chung, K.Y.; Shin, D.H.; Kim, Y.S.; Kim, S.K.; Kim, S.K. Identification of Tumor Suppressor Loci on the Long Arm of Chromosome 15 in Primary Small Cell Lung Cancer. Yonsei Med. J. 2003, 44, 65. [Google Scholar] [CrossRef]
- Kaur, E.; Agrawal, R.; Sengupta, S. Functions of BLM Helicase in Cells: Is It Acting Like a Double-Edged Sword? Front. Genet. 2021, 12, 634789. [Google Scholar] [CrossRef]
- Yuan, X.-W.; Wang, D.-M.; Hu, Y.; Tang, Y.-N.; Shi, W.-W.; Guo, X.-J.; Song, J.-G. Hepatocyte Nuclear Factor 6 Suppresses the Migration and Invasive Growth of Lung Cancer Cells through P53 and the Inhibition of Epithelial-Mesenchymal Transition. J. Biol. Chem. 2013, 288, 31206–31216. [Google Scholar] [CrossRef]
- Gurrieri, C.; Capodieci, P.; Bernardi, R.; Scaglioni, P.P.; Nafa, K.; Rush, L.J.; Verbel, D.A.; Cordon-Cardo, C.; Pandolfi, P.P. Loss of the Tumor Suppressor PML in Human Cancers of Multiple Histologic Origins. J. Natl. Cancer Inst. 2004, 96, 269–279. [Google Scholar] [CrossRef]
- Huang, T.; Sun, L.; Yuan, X.; Qiu, H. Thrombospondin-1 Is a Multifaceted Player in Tumor Progression. Oncotarget 2017, 8, 84546–84558. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, B.; Moran, M.S.; Zhao, Y.; Su, P.; Haffty, B.G.; Shao, C.; Yang, Q. 53BP1 Functions as a Tumor Suppressor in Breast Cancer via the Inhibition of NF-ΚB through MiR-146a. Carcinogenesis 2012, 33, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Parrish, A.B.; Wright, K.M.; Margolis, S.S.; Marks, J.R.; Deshmukh, M.; Kornbluth, S. Enhanced Sensitivity to Cytochrome c –Induced Apoptosis Mediated by PHAPI in Breast Cancer Cells. Cancer Res. 2006, 66, 2210–2218. [Google Scholar] [CrossRef]
- Wu, S.; Xue, W.; Huang, X.; Yu, X.; Luo, M.; Huang, Y.; Liu, Y.; Bi, Z.; Qiu, X.; Bai, S. Distinct Prognostic Values of ALDH1 Isoenzymes in Breast Cancer. Tumor Biol. 2015, 36, 2421–2426. [Google Scholar] [CrossRef]
- Ma, W.; Stafford, L.J.; Li, D.; Luo, J.; Li, X.; Ning, G.; Liu, M. GCIP/CCNDBP1, a Helix–Loop–Helix Protein, Suppresses Tumorigenesis. J. Cell. Biochem. 2007, 100, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Oudenaarden, C.R.L.; van de Ven, R.A.H.; Derksen, P.W.B. Re-Inforcing the Cell Death Army in the Fight against Breast Cancer. J. Cell Sci. 2018, 131, jcs212563. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.E.; Kim, Y.W.; Hwang, S.Y.; Shin, S.M.; Shin, J.W.; Lee, Y.H.; Shin, S.Y.; Han, K.T.; Lee, J.M.; Namkoong, S.E.; et al. Candidate Tumor Suppressor, HCCS-1, Is Downregulated in Human Cancers and Induces Apoptosis in Cervical Cancer. Int. J. Cancer 2002, 97, 780–786. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Z.; Yi, Z.; Li, J. MicroRNA-211-5p Suppresses Tumour Cell Proliferation, Invasion, Migration and Metastasis in Triple-Negative Breast Cancer by Directly Targeting SETBP1. Br. J. Cancer 2017, 117, 78–88. [Google Scholar] [CrossRef]
- Li, M.; Pan, M.; Wang, J.; You, C.; Zhao, F.; Zheng, D.; Guo, M.; Xu, H.; Wu, D.; Wang, L.; et al. MiR-7 Reduces Breast Cancer Stem Cell Metastasis via Inhibiting RELA to Decrease ESAM Expression. Mol. Ther.-Oncolytics 2020, 18, 70–82. [Google Scholar] [CrossRef]
- Bandini, E.; Fanini, F.; Vannini, I.; Rossi, T.; Plousiou, M.; Tumedei, M.M.; Limarzi, F.; Maltoni, R.; Fabbri, F.; Hrelia, S.; et al. MiR-9-5p as a Regulator of the Androgen Receptor Pathway in Breast Cancer Cell Lines. Front. Cell Dev. Biol. 2020, 8, 579160. [Google Scholar] [CrossRef] [PubMed]
- Barbano, R.; Pasculli, B.; Rendina, M.; Fontana, A.; Fusilli, C.; Copetti, M.; Castellana, S.; Valori, V.M.; Morritti, M.; Graziano, P.; et al. Stepwise Analysis of MIR9 Loci Identifies MiR-9-5p to Be Involved in Oestrogen Regulated Pathways in Breast Cancer Patients. Sci. Rep. 2017, 7, 45283. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Chen, Y.; Yao, S.; Deng, G.; Liu, D.; Yuan, X.; Liu, S.; Rao, J.; Xiong, H.; Yuan, X.; et al. MiR-422a Weakened Breast Cancer Stem Cells Properties by Targeting PLP2. Cancer Biol. Ther. 2018, 19, 436–444. [Google Scholar] [CrossRef]
- Silvestri, M.; Dugo, M.; Vismara, M.; De Cecco, L.; Lanzoni, D.; Vingiani, A.; Folli, S.; De Santis, M.C.; de Braud, F.; Pruneri, G.; et al. Copy Number Alterations Analysis of Primary Tumor Tissue and Circulating Tumor Cells from Patients with Early-Stage Triple Negative Breast Cancer. Sci. Rep. 2022, 12, 1470. [Google Scholar] [CrossRef] [PubMed]
- Menyailo, M.E.; Tretyakova, M.S.; Denisov, E. V Heterogeneity of Circulating Tumor Cells in Breast Cancer: Identifying Metastatic Seeds. Int. J. Mol. Sci. 2020, 21, 1696. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.M.M.; et al. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Riebensahm, C.; Joosse, S.A.; Mohme, M.; Hanssen, A.; Matschke, J.; Goy, Y.; Witzel, I.; Lamszus, K.; Kropidlowski, J.; Petersen, C.; et al. Clonality of Circulating Tumor Cells in Breast Cancer Brain Metastasis Patients. Breast Cancer Res. 2019, 21, 101. [Google Scholar] [CrossRef] [PubMed]
- Fusco, N.; Malapelle, U.; Fassan, M.; Marchiò, C.; Buglioni, S.; Zupo, S.; Criscitiello, C.; Vigneri, P.; Dei Tos, A.P.; Maiorano, E.; et al. PIK3CA Mutations as a Molecular Target for Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Front. Oncol. 2021, 11, 644737. [Google Scholar] [CrossRef] [PubMed]
- Pestrin, M.; Salvianti, F.; Galardi, F.; de Luca, F.; Turner, N.; Malorni, L.; Pazzagli, M.; di Leo, A.; Pinzani, P. Heterogeneity of PIK3CA Mutational Status at the Single Cell Level in Circulating Tumor Cells from Metastatic Breast Cancer Patients. Mol. Oncol. 2015, 9, 749–757. [Google Scholar] [CrossRef]
- Deng, G.; Krishnakumar, S.; Powell, A.A.; Zhang, H.; Mindrinos, M.N.; Telli, M.L.; Davis, R.W.; Jeffrey, S.S. Single Cell Mutational Analysis of PIK3CA in Circulating Tumor Cells and Metastases in Breast Cancer Reveals Heterogeneity, Discordance, and Mutation Persistence in Cultured Disseminated Tumor Cells from Bone Marrow. BMC Cancer 2014, 14, 456. [Google Scholar] [CrossRef]
- Rossi, T.; Palleschi, M.; Angeli, D.; Tebaldi, M.; Martinelli, G.; Vannini, I.; Puccetti, M.; Limarzi, F.; Maltoni, R.; Gallerani, G.; et al. Case Report: Analysis of Circulating Tumor Cells in a Triple Negative Spindle-Cell Metaplastic Breast Cancer Patient. Front. Med. 2021, 8, 856. [Google Scholar] [CrossRef] [PubMed]
- D’Oronzo, S.; Lovero, D.; Palmirotta, R.; Stucci, L.S.; Tucci, M.; Felici, C.; Cascardi, E.; Giardina, C.; Cafforio, P.; Silvestris, F. Dissection of Major Cancer Gene Variants in Subsets of Circulating Tumor Cells in Advanced Breast Cancer. Sci. Rep. 2019, 9, 17276. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Miyoshi, Y. Mechanism of Resistance to Endocrine Therapy in Breast Cancer: The Important Role of PI3K/Akt/MTOR in Estrogen Receptor-Positive, HER2-Negative Breast Cancer. Breast Cancer 2018, 25, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Gallerani, G.; Rossi, T.; Valgiusti, M.; Angeli, D.; Fici, P.; De Fanti, S.; Bandini, E.; Cocchi, C.; Frassineti, G.L.; Bonafè, M.; et al. CNA Profiling of Single CTCs in Locally Advanced Esophageal Cancer Patients during Therapy Highlights Unexplored Molecular Pathways. Cancers 2021, 13, 6369. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-C.W.; Lopez-Diaz, F.J.; Khan, S.Y.; Tariq, M.A.; Dayn, Y.; Vaske, C.J.; Radenbaugh, A.J.; Kim, H.J.; Emerson, B.M.; Pourmand, N. Single-Cell Analyses of Transcriptional Heterogeneity during Drug Tolerance Transition in Cancer Cells by RNA Sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, E4726–E4735. [Google Scholar] [CrossRef] [PubMed]
- WANG, S.-H.; LI, N.; WEI, Y.; LI, Q.-R.; YU, Z.-P. β-Catenin Deacetylation Is Essential for WNT-Induced Proliferation of Breast Cancer Cells. Mol. Med. Rep. 2014, 9, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.N.; Hedditch, E.L.; Baker, L.A.; Jary, E.; Ward, R.L.; Ford, C.E. The Wnt Signalling Pathway Is Upregulated in an in Vitro Model of Acquired Tamoxifen Resistant Breast Cancer. BMC Cancer 2013, 13, 174. [Google Scholar] [CrossRef] [PubMed]
- Bostner, J.; Ahnström Waltersson, M.; Fornander, T.; Skoog, L.; Nordenskjöld, B.; Stål, O. Amplification of CCND1 and PAK1 as Predictors of Recurrence and Tamoxifen Resistance in Postmenopausal Breast Cancer. Oncogene 2007, 26, 6997–7005. [Google Scholar] [CrossRef]
- Butt, A.J.; McNeil, C.M.; Musgrove, E.A.; Sutherland, R.L. Downstream Targets of Growth Factor and Oestrogen Signalling and Endocrine Resistance: The Potential Roles of c-Myc, Cyclin D1 and Cyclin E. Endocr.-Relat. Cancer 2005, 12, S47–S59. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.G.; Pratt, N.; Purdie, C.A.; Baker, L.; Ashfield, A.; Quinlan, P.; Thompson, A.M. High CCND1 Amplification Identifies a Group of Poor Prognosis Women with Estrogen Receptor Positive Breast Cancer. Int. J. Cancer 2010, 127, 355–360. [Google Scholar] [CrossRef]
- Tchakarska, G.; Sola, B. The Double Dealing of Cyclin D1. Cell Cycle 2020, 19, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Massink, M.P.G.; Kooi, I.E.; Martens, J.W.M.; Waisfisz, Q.; Meijers-Heijboer, H. Genomic Profiling of CHEK2*1100delC-Mutated Breast Carcinomas. BMC Cancer 2015, 15, 877. [Google Scholar] [CrossRef] [PubMed]
- Nevanlinna, H.; Bartek, J. The CHEK2 Gene and Inherited Breast Cancer Susceptibility. Oncogene 2006, 25, 5912–5919. [Google Scholar] [CrossRef] [PubMed]
- Castro-Giner, F.; Aceto, N. Tracking Cancer Progression: From Circulating Tumor Cells to Metastasis. Genome Med. 2020, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, T.V.; Gorbunova, A.S.; Zhivotovsky, B. Mitochondrial Involvement in Migration, Invasion and Metastasis. Front. Cell Dev. Biol. 2019, 7, 355. [Google Scholar] [CrossRef]
- Si, M.; Lang, J. The Roles of Metallothioneins in Carcinogenesis. J. Hematol. Oncol. 2018, 11, 107. [Google Scholar] [CrossRef]
- Natrajan, R.; Louhelainen, J.; Williams, S.; Laye, J.; Knowles, M.A. High-Resolution Deletion Mapping of 15q13.2-Q21.1 in Transitional Cell Carcinoma of the Bladder. Cancer Res. 2003, 63, 7657–7662. [Google Scholar]
- Fang, M.; Toher, J.; Morgan, M.; Davison, J.; Tannenbaum, S.; Claffey, K. Genomic Differences between Estrogen Receptor (ER)-Positive and ER-Negative Human Breast Carcinoma Identified by Single Nucleotide Polymorphism Array Comparative Genome Hybridization Analysis. Cancer 2011, 117, 2024–2034. [Google Scholar] [CrossRef]
- Shankar, J.; Messenberg, A.; Chan, J.; Underhill, T.M.; Foster, L.J.; Nabi, I.R. Pseudopodial Actin Dynamics Control Epithelial-Mesenchymal Transition in Metastatic Cancer Cells. Cancer Res. 2010, 70, 3780–3790. [Google Scholar] [CrossRef]
- Choi, S.; Bhagwat, A.M.; Al Mismar, R.; Goswami, N.; Ben Hamidane, H.; Sun, L.; Graumann, J. Proteomic Profiling of Human Cancer Pseudopodia for the Identification of Anti-Metastatic Drug Candidates. Sci. Rep. 2018, 8, 5858. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Therapy | Clinical Response | Site of Metastasis | Number of CTC Isolated | |||
|---|---|---|---|---|---|---|---|
| Timepoint A | Timepoint B | Timepoint C | Total | ||||
| CH28 | Capecitabine | PR | Bone, Lymph nodes | 5 | 5 | ||
| CH29 | Fulvestrant + Palbociclib | PD | Peritoneum | 17 | 9 | 30 | 56 |
| CH30 | Capecitabine + Vinorelbine | PD | Bone, Liver | 37 | 2 | 39 | |
| CH32 | Capecitabine + Vinorelbine | SD | Bone, Liver, Lung | 15 | 15 | ||
| CH46 | Letrozole + Ribociclib | SD | Bone | 5 | 5 | ||
| CH47 | Letrozole + Ribociclib | N/A | Bone, Lymph nodes | 4 | 4 | ||
| Total | 83 | 9 | 32 | 124 | |||
| Gene | Gene Product | Genomic Location | References |
|---|---|---|---|
| BLM | Bloom Syndrome RecQ Like Helicase | 15q26.1 | [38] |
| ONECUT1 | One Cut Homeobox 1 | 15q21.3 | [39] |
| PML | Promyelocytic Leukemia Protein | 15q22 | [40] |
| THBS1 | Thrombospondin-1 | 15q15 | [41] |
| TP53BP1 | Tumor Protein P53 Binding Protein 1 | 15q15-q21 | [42] |
| ANP32A | Acidic Nuclear Phosphoprotein 32 Family Member A | 15q23 | [43] |
| ALDH1A2 | Aldehyde Dehydrogenase 1 Family Member A2 | 15q21.3 | [44] |
| CCNDBP1 | Cyclin D1 Binding Protein 1 | 15q14-q15 | [45] |
| BMF | Bcl2 Modifying Factor | 15q14 | [46] |
| ST20 | Suppressor Of Tumorigenicity 20 | 15q25.1 | [47] |
| MIR211 | Hsa-Mir-211 | 15q13.3 | [48] |
| MIR7-2 | Hsa-Mir-7 | 15q26.1 | [49] |
| MIR9-3 | Hsa-Mir-9 | 15q26.1 | [50,51] |
| MIR422A | Hsa-Mir-422a | 15q22.31 | [52] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, T.; Angeli, D.; Tebaldi, M.; Fici, P.; Rossi, E.; Rocca, A.; Palleschi, M.; Maltoni, R.; Martinelli, G.; Fabbri, F.; et al. Dissecting Molecular Heterogeneity of Circulating Tumor Cells (CTCs) from Metastatic Breast Cancer Patients through Copy Number Aberration (CNA) and Single Nucleotide Variant (SNV) Single Cell Analysis. Cancers 2022, 14, 3925. https://doi.org/10.3390/cancers14163925
Rossi T, Angeli D, Tebaldi M, Fici P, Rossi E, Rocca A, Palleschi M, Maltoni R, Martinelli G, Fabbri F, et al. Dissecting Molecular Heterogeneity of Circulating Tumor Cells (CTCs) from Metastatic Breast Cancer Patients through Copy Number Aberration (CNA) and Single Nucleotide Variant (SNV) Single Cell Analysis. Cancers. 2022; 14(16):3925. https://doi.org/10.3390/cancers14163925
Chicago/Turabian StyleRossi, Tania, Davide Angeli, Michela Tebaldi, Pietro Fici, Elisabetta Rossi, Andrea Rocca, Michela Palleschi, Roberta Maltoni, Giovanni Martinelli, Francesco Fabbri, and et al. 2022. "Dissecting Molecular Heterogeneity of Circulating Tumor Cells (CTCs) from Metastatic Breast Cancer Patients through Copy Number Aberration (CNA) and Single Nucleotide Variant (SNV) Single Cell Analysis" Cancers 14, no. 16: 3925. https://doi.org/10.3390/cancers14163925
APA StyleRossi, T., Angeli, D., Tebaldi, M., Fici, P., Rossi, E., Rocca, A., Palleschi, M., Maltoni, R., Martinelli, G., Fabbri, F., & Gallerani, G. (2022). Dissecting Molecular Heterogeneity of Circulating Tumor Cells (CTCs) from Metastatic Breast Cancer Patients through Copy Number Aberration (CNA) and Single Nucleotide Variant (SNV) Single Cell Analysis. Cancers, 14(16), 3925. https://doi.org/10.3390/cancers14163925