Tumor Antigenicity and a Pre-Existing Adaptive Immune Response in Advanced BRAF Mutant Colorectal Cancers

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Clinical-Pathologic Study
2.2. RNA Extraction and Hybridization to nCounter Codeset
2.3. Statistical Analysis of NanoString PanCancer IO 360 Panel Data
2.4. Statistical Analysis of Clinico-Pathological Data
3. Results
3.1. MMR Status, CD8+ TIL Infiltrate and Clinical-Pathological Features of BRAF-CRCs
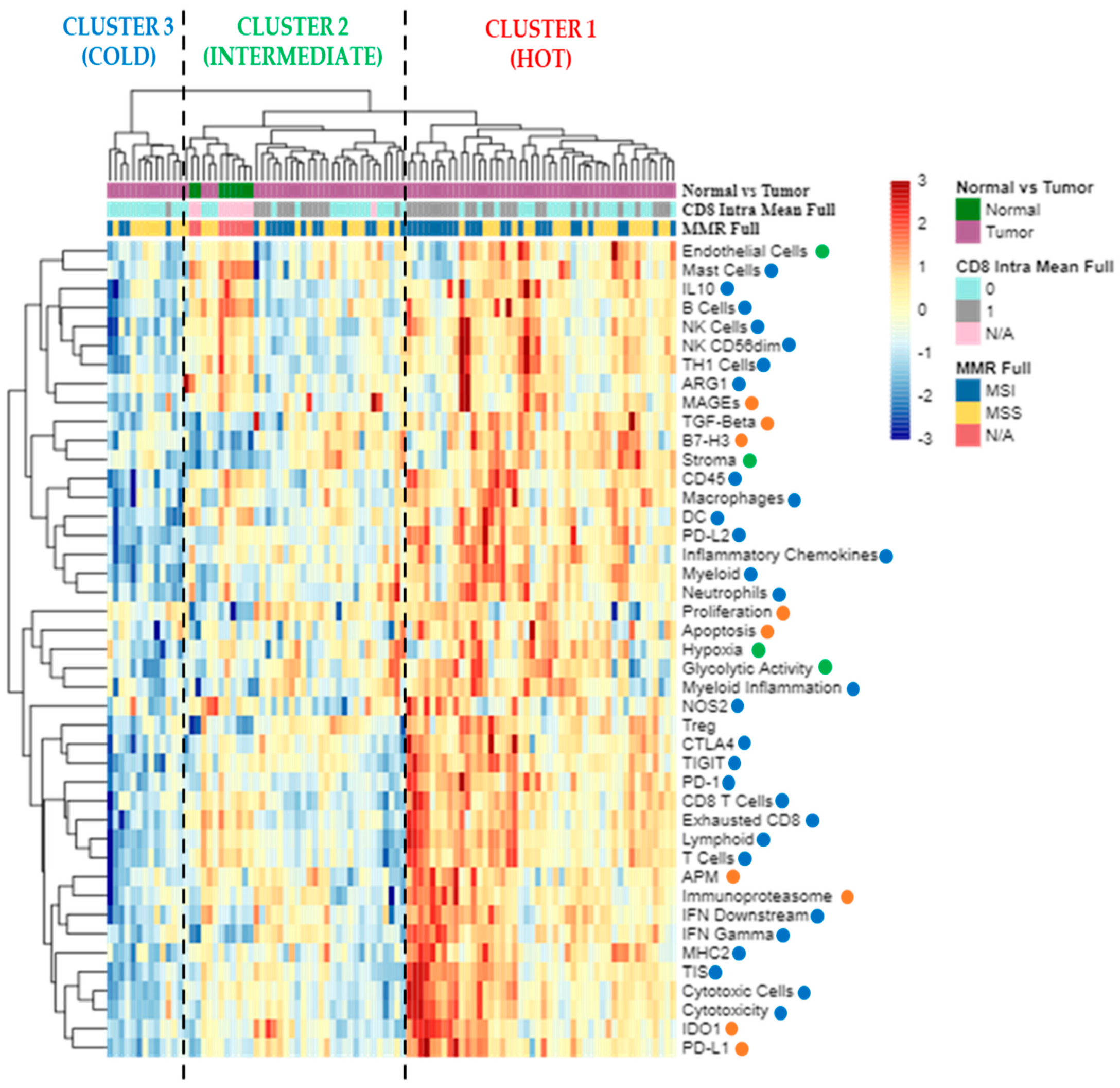
3.2. High and Low Immune Profiles of BRAF-CRCs
3.3. Differential Gene Expression in MSI vs. MSS and in CD8+ vs. CD8− BRAF-CRCs
3.4. Survival Analysis
3.5. Tumor Inflammation Signature (TIS) within BRAF-CRCs and Correlation with CD8+ TIL Content and with MSI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Y.C.; Gotea, V.; Margolin, G.; Elnitski, L. Significant associations between driver gene mutations and DNA methylation alterations across many cancer types. PLoS Comput. Biol. 2017, 13, e1005840. [Google Scholar] [CrossRef] [PubMed]
- Thiel, A.; Heinonen, M.; Kantonen, J.; Gylling, A.; Lahtinen, L.; Korhonen, M.; Kytölä, S.; Mecklin, J.P.; Orpana, A.; Peltomäki, P.; et al. BRAF mutation in sporadic colorectal cancer and Lynch syndrome. Virchows Arch. 2013, 463, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Garcia, E.; Argiles, G.; Elez, E.; Tabernero, J. BRAF mutant colorectal cancer: Prognosis, treatment, and new perspectives. Ann. Oncol. 2017, 28, 2648–2657. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Chalabi, M.; Fanchi, L.F.; Dijkstra, K.K.; Van den Berg, J.G.; Aalbers, A.G.; Sikorska, K.; Lopez-Yurda, M.; Grootscholten, C.; Beets, G.L.; Snaebjornsson, P.; et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020, 26, 566–576. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Ludford, K.; Cohen, R.; Svrcek, M.; Foo, W.C.; Colle, R.; Parc, Y.; Thomas, J.V.; Morris, V.K.; Kopetz, S.; Chang, G.J.; et al. Pathological Tumor Response Following Immune Checkpoint Blockade for Deficient Mismatch Repair Advanced Colorectal Cancer. J. Natl. Cancer Inst. 2021, 113, 208–211. [Google Scholar] [CrossRef]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Rozeman, E.A.; Fanchi, L.F.; Sikorska, K.; van de Wiel, B.; Kvistborg, P.; Krijgsman, O.; van den Braber, M.; Philips, D.; Broeks, A.; et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 2018, 24, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Amaria, R.N.; Reddy, S.M.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Orlowski, R.J.; Xu, X.; Mick, R.; George, S.M.; Yan, P.K.; Manne, S.; Kraya, A.A.; Wubbenhorst, B.; Dorfman, L.; et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med. 2019, 25, 454–461. [Google Scholar] [CrossRef]
- Powles, T.; Kockx, M.; Rodriguez-Vida, A.; Duran, I.; Crabb, S.J.; Van Der Heijden, M.S.; Szabados, B.; Pous, A.F.; Gravis, G.; Herranz, U.A.; et al. Clinical efficacy and biomarker analysis of neoadjuvant atezolizumab in operable urothelial carcinoma in the ABACUS trial. Nat. Med. 2019, 25, 1706–1714. [Google Scholar] [CrossRef]
- Barras, D.; Missiaglia, E.; Wirapati, P.; Sieber, O.M.; Jorissen, R.N.; Love, C.; Molloy, P.L.; Jones, I.T.; McLaughlin, S.; Gibbs, P.; et al. BRAF V600E Mutant Colorectal Cancer Subtypes Based on Gene Expression. Clin. Cancer Res. 2017, 23, 104–115. [Google Scholar] [CrossRef]
- Digiacomo, N.; Bolzacchini, E.; Veronesi, G.; Cerutti, R.; Sahnane, N.; Pinotti, G.; Bregni, M.; Artale, S.; Verusio, C.; Crivelli, F.; et al. Neuroendocrine Differentiation, Microsatellite Instability, and Tumor-infiltrating Lymphocytes in Advanced Colorectal Cancer With BRAF Mutation. Clin. Colorectal Cancer 2019, 18, e251–e260. [Google Scholar] [CrossRef]
- Cristescu, R.; Nebozhyn, M.; Zhang, C.; Albright, A.; Kobie, J.; Huang, L.; Zhao, Q.; Wang, A.; Ma, H.; Alexander Cao, Z.; et al. Transcriptomic Determinants of Response to Pembrolizumab Monotherapy across Solid Tumor Types. Clin. Cancer Res. 2022, 28, 1680–1689. [Google Scholar] [CrossRef]
- Haddad, R.I.; Seiwert, T.Y.; Chow, L.Q.M.; Gupta, S.; Weiss, J.; Gluck, I.; Eder, J.P.; Burtness, B.; Tahara, M.; Keam, B.; et al. Influence of tumor mutational burden, inflammatory gene expression profile, and PD-L1 expression on response to pembrolizumab in head and neck squamous cell carcinoma. J. Immunother. Cancer 2022, 10, e003026. [Google Scholar] [CrossRef]
- Damotte, D.; Warren, S.; Arrondeau, J.; Boudou-Rouquette, P.; Mansuet-Lupo, A.; Biton, J.; Ouakrim, H.; Alifano, M.; Gervais, C.; Bellesoeur, A.; et al. The tumor inflammation signature (TIS) is associated with anti-PD-1 treatment benefit in the CERTIM pan-cancer cohort. J. Transl. Med. 2019, 17, 357. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Danaher, P.; Warren, S.; Lu, R.; Samayoa, J.; Sullivan, A.; Pekker, I.; Wallden, B.; Marincola, F.M.; Cesano, A. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): Results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 2018, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Csiba, A.; Whitwell, H.L.; Moore, M. Distribution of histocompatibility and leucocyte differentiation antigens in normal human colon and in benign and malignant colonic neoplasms. Br. J. Cancer 1984, 50, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Momburg, F.; Degener, T.; Bacchus, E.; Moldenhauer, G.; Hämmerling, G.J.; Möller, P. Loss of HLA-A,B,C and de novo expression of HLA-D in colorectal cancer. Int. J. Cancer 1986, 37, 179–184. [Google Scholar] [CrossRef]
- Moore, M.; Ghosh, A.K.; Johnston, D.; Street, A.J. Expression of MHC class II products on human colorectal cancer. An immunohistological and flow cytometric study. J. Immunogenet. 1986, 13, 201–209. [Google Scholar] [CrossRef]
- Durrant, L.G.; Ballantyne, K.C.; Armitage, N.C.; Robins, R.A.; Marksman, R.; Hardcastle, J.D.; Baldwin, R.W. Quantitation of MHC antigen expression on colorectal tumours and its association with tumour progression. Br. J. Cancer 1987, 56, 425–432. [Google Scholar] [CrossRef]
- Rees, R.C.; Buckle, A.M.; Gelsthorpe, K.; James, V.; Potter, C.W.; Rogers, K.; Jacob, G. Loss of polymorphic A and B locus HLA antigens in colon carcinoma. Br. J. Cancer 1988, 57, 374–377. [Google Scholar] [CrossRef]
- Smith, M.E.; Marsh, S.G.; Bodmer, J.G.; Gelsthorpe, K.; Bodmer, W.F. Loss of HLA-A,B,C allele products and lymphocyte function-associated antigen 3 in colorectal neoplasia. Proc. Natl. Acad. Sci. USA 1989, 86, 5557–5561. [Google Scholar] [CrossRef]
- López-Nevot, M.A.; Esteban, F.; Ferrón, A.; Gutiérrez, J.; Oliva, M.R.; Romero, C.; Huelin, C.; Ruiz-Cabello, F.; Garrido, F. HLA class I gene expression on human primary tumours and autologous metastases: Demonstration of selective losses of HLA antigens on colorectal, gastric and laryngeal carcinomas. Br. J. Cancer 1989, 59, 221–226. [Google Scholar] [CrossRef]
- Anderson, P.; Aptsiauri, N.; Ruiz-Cabello, F.; Garrido, F. HLA class I loss in colorectal cancer: Implications for immune escape and immunotherapy. Cell. Mol. Immunol. 2021, 18, 556–565. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Cabrera, T.; Aptsiauri, N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int. J. Cancer 2010, 127, 249–256. [Google Scholar] [CrossRef]
- Rodríguez, T.; Méndez, R.; Del Campo, A.; Jiménez, P.; Aptsiauri, N.; Garrido, F.; Ruiz-Cabello, F. Distinct mechanisms of loss of IFN-gamma mediated HLA class I inducibility in two melanoma cell lines. BMC Cancer 2007, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Carretero, R.; Romero, J.M.; Ruiz-Cabello, F.; Maleno, I.; Rodriguez, F.; Camacho, F.M.; Real, L.M.; Garrido, F.; Cabrera, T. Analysis of HLA class I expression in progressing and regressing metastatic melanoma lesions after immunotherapy. Immunogenetics 2008, 60, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Kloor, M.; Michel, S.; Buckowitz, B.; Rüschoff, J.; Büttner, R.; Holinski-Feder, E.; Dippold, W.; Wagner, R.; Tariverdian, M.; Benner, A.; et al. Beta2-microglobulin mutations in microsatellite unstable colorectal tumors. Int. J. Cancer 2007, 121, 454–458. [Google Scholar] [CrossRef]
- Bernal, M.; García-Alcalde, F.; Concha, A.; Cano, C.; Blanco, A.; Garrido, F.; Ruiz-Cabello, F. Genome-wide differential genetic profiling characterizes colorectal cancers with genetic instability and specific routes to HLA class I loss and immune escape. Cancer Immunol. Immunother. 2012, 61, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Bernal, M.; Ruiz-Cabello, F.; Concha, A.; Paschen, A.; Garrido, F. Implication of the β2-microglobulin gene in the generation of tumor escape phenotypes. Cancer Immunol. Immunother. 2012, 61, 1359–1371. [Google Scholar] [CrossRef]
- Bicknell, D.C.; Kaklamanis, L.; Hampson, R.; Bodmer, W.F.; Karran, P. Selection for beta 2-microglobulin mutation in mismatch repair-defective colorectal carcinomas. Curr. Biol. 1996, 6, 1695–1697. [Google Scholar] [CrossRef]
- Kloor, M.; Becker, C.; Benner, A.; Woerner, S.M.; Gebert, J.; Ferrone, S.; von Knebel Doeberitz, M. Immunoselective pressure and human leukocyte antigen class I antigen machinery defects in microsatellite unstable colorectal cancers. Cancer Res. 2005, 65, 6418–6424. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | All Cases § (n = 89) | MSI CRC (n = 43) | MSS CRC (n = 46) | p Value | CD8+ CRC # (n = 39) | CD8- CRC # (n = 49) | p Value |
|---|---|---|---|---|---|---|---|
| MMR (MSI) | 43 (48%) | - | - | - | 27 (69%) | 15 (31%) | 0.0001 |
| CD8+ (n = 88) | 39 (44%) | 27 (64%) | 12 (26%) | 0.0001 | - | - | - |
| Age, y (mean ± SD) | 69.8 ± 10.1 | 72.1 ± 8.6 | 67.7 ± 11.1 | 0.0311 | 72.1 ± 8.8 | 68.1 ± 11 | ns (0.07) |
| Female sex | 54 (61%) | 28 (65%) | 26 (57%) | ns | 22 (56%) | 31 (63%) | ns |
| Right tumor site | 64 (73%) | 39 (91%) | 25 (56%) | <0.0001 | 31 (79%) | 32 (67%) | ns |
| Histologic type | 0.004 | 0.005 | |||||
| Tubular | 38 (43%) | 12 (28%) | 26 (57%) | 10 (26%) | 27 (55%) | ||
| Mucinous/signet ring cells | 33 (37%) | 21 (49%) | 12 (25%) | 20 (51%) | 13 (27%) | ||
| Medullary/undifferentiated | 12 (13%) | 9 (21%) | 3 (7%) | 8 (21%) | 4 (8%) | ||
| MANEC | 6 (7%) | 1 (2%) | 5 (11%) | 1 (3%) | 5 (10%) | ||
| High Grade | 43 (48%) | 27 (63%) | 16 (35%) | 0.008 | 23 (59%) | 19 (39%) | ns (0.08) |
| Stage at diagnosis (I-III) | 44 (49%) | 27 (63%) | 17 (37%) | 0.015 | 22 (56%) | 21 (43%) | ns |
| Distant Metastases (n = 79) | 0.0013 | 0.0291 | |||||
| Presence * | 77 (86,5%) | 32 (74%) | 45 (98%) | 30 (77%) | 46 (94%) | ||
| Absence | 12 (13,5%) | 11 (26%) | 1 (2%) | 9 (23%) | 3 (6%) | ||
| Infiltrative Growth pattern (n = 83) | 64 (77%) | 30 (70%) | 40 (87%) | ns | 27 (69%) | 42 (86%) | ns |
| Stroma ≥ 20% (n = 87) | 52 (60%) | 18 (43%) | 34 (76%) | 0.0023 | 13 (34%) | 21 (44%) | ns |
| Synaptophysin ≥ 1 (n = 77) | 16 (21%) | 1 (3%) | 15 (39%) | 0.0001 | 1 (3%) | 15 (33%) | 0.001 |
| p53 ≥ 50 (n = 67) | 22 (34%) | 2 (7%) | 20 (51%) | 0.0002 | 3 (14%) | 19 (43%) | 0.0255 |
| PD-L1 ≥ 1 (n = 65) | 17 (26%) | 16 (94%) | 1 (6%) | <0.0001 | 14 (82%) | 3 (18%) | <0.0001 |
| Tumor Signature | Tumor Microenvironment Signature | Immune Response Signature | |||||
|---|---|---|---|---|---|---|---|
| Tumor Immunogenicity | Tumor Sensitivity to Immune Attack | Inhibitory Tumor Mechanisms | Stromal Factors | Inhibitory Metabolism | Inhibitory Immune Signaling | Anti-TumorImmune Activity | Immune Cell Population Abundance |
| APM | Apoptosis | B7-H3 | Endothelial Cells | Glycol Act | ARG1 | TIS | B Cells |
| APM Loss | JAK-STAT Loss | IDO1 | Stroma | Hypoxia | CTLA4 | Cytotoxicity | CD45 |
| Hypermutation | Proliferation | PD-L1 | IL10 | IFN Down | CD8 T Cells | ||
| Immunoproteasome | TGF-Beta | Inflam Chemokine | IFN Gamma | Cytotoxic Cells | |||
| MAGEs | Myeloid Inflam | Lymphoid | DC | ||||
| MMR Loss | NOS2 | MHC2 | Exhausted CD8 | ||||
| MSI Predictor | PD-1 | Myeloid | Macrophages | ||||
| PD-L2 | Mast Cells | ||||||
| TIGIT | Neutrophils | ||||||
| NK CD56dim | |||||||
| NK Cells | |||||||
| T Cells | |||||||
| TH1 Cells | |||||||
| Treg | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolzacchini, E.; Libera, L.; Church, S.E.; Sahnane, N.; Bombelli, R.; Digiacomo, N.; Giordano, M.; Petracco, G.; Sessa, F.; Capella, C.; et al. Tumor Antigenicity and a Pre-Existing Adaptive Immune Response in Advanced BRAF Mutant Colorectal Cancers. Cancers 2022, 14, 3951. https://doi.org/10.3390/cancers14163951
Bolzacchini E, Libera L, Church SE, Sahnane N, Bombelli R, Digiacomo N, Giordano M, Petracco G, Sessa F, Capella C, et al. Tumor Antigenicity and a Pre-Existing Adaptive Immune Response in Advanced BRAF Mutant Colorectal Cancers. Cancers. 2022; 14(16):3951. https://doi.org/10.3390/cancers14163951
Chicago/Turabian StyleBolzacchini, Elena, Laura Libera, Sarah E. Church, Nora Sahnane, Raffaella Bombelli, Nunzio Digiacomo, Monica Giordano, Guido Petracco, Fausto Sessa, Carlo Capella, and et al. 2022. "Tumor Antigenicity and a Pre-Existing Adaptive Immune Response in Advanced BRAF Mutant Colorectal Cancers" Cancers 14, no. 16: 3951. https://doi.org/10.3390/cancers14163951